
Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine1
Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disorder associated with a global impairment of higher mental function, and presenting an impairment of memory as the cardinal symptom[1]. Histopathological hallmarks of the disease are the extracellular deposition of amyloid β-peptide (Aβ) in senile plaques, the appearance of intracellular neurofibrillary tangles (NFT), a loss of cholinergic neurons, and extensive synaptic changes in the cerebral cortex, hippo-campus and other areas of brain essential for cognitive functions. The key symptoms of AD are primarily caused by cholinergic dysfunction. A significant correlation has been found between a decrease in cortical cholinergic activity and the deterioration of mental test scores in patients with AD[2]. Based on the cholinergic hypothesis of AD, cholinergic enhancement strategies have been at the forefront of efforts to pharmacologically palliate the cognitive impairments. Among the various therapeutic approaches investigated to enhance cholinergic transmission, cholinesterase inhibitors (ChEI) are the first group of compounds that have shown some promise in the treatment of AD. The most significant therapeutic effect of ChEI in AD treatment is to stabilize cognitive function at steady level during at least a 1 year period in approximately 50% of patients. Most clinical studies show that in a certain percentage of AD patients (approximately 20%) cognitive function can be stabilized for a period of up to 24 months. In addition, the AD patients who do not respond to therapy with one ChEI can be switched to a second one with a 50% rate of success. To date, four ChEI, tacrine, donepezil, galanthamine and rivastigmine have been approved by the US Food and Drug Administration for the treatment of AD, and several new ChEI are being studied[3–6]. However, the clinical usefulness of ChEI has been limited by their short half-lives and excessive side effects caused by activation of peripheral cholinergic systems, as well as hepatotoxicity, which is the most frequent and important side effect of tacrine therapy[7–9]. To obtain better therapeutic benefit in the treatment of AD, the search for a long-acting ChEI that exerts minimal clinical side effects is still ongoing[5].
(–)-Huperzine A (HupA), a novel Lycopodium alkaloid, is isolated from the Chinese medicinal herb Huperzia serrata (Qian Ceng Ta; Figure 1). The herb has been used in China for centuries in the treatment of such conditions as contu-sions, strains, swelling, and schizophrenia. HupA, a compound that is chemically unique in comparison with other agents under study for AD, is a reversible, potent, and selective acetylcholinesterase (AChE) inhibitor. Its potency and duration of AChE inhibition rival those of tacrine, galantha-mine, donepezil, and rivastigmine[10–13]. HupA has been found to improve cognitive deficits in a broad range of animal models. The phase IV clinical trials conducted in China have demonstrated that HupA induces significant improvement in the memory of elderly people and patients with AD and vascular dementia (VD) without any notable side effects[14–17]. In this paper the pharmacological properties, pharmacokine-tics, and toxicology of HupA, in addition to the clinical trials so far conducted using this agent, are reviewed.

Effects on cholinesterase activity and inhibition mechanism
The cholinesterase inhibition by HupA has been evaluated in vitro and in vivo, using a spectrophotometric me-thod[18] with minor modifications. For assay of AChE or butyrylcholinesterase (BuChE) activity, a reaction mixture of 4 mL containing acetylthiocholine iodide (0.3 mmol/L) or butyrylthiocholine iodide (0.4 mmol/L), 1 mL sodium phosphate buffer (0.1 mmol/L), the test compound (0.1–0.5 mL), and enzyme (0.1–0.2 mL) was incubated at 37 °C for 8 min.
In vitro and in vivo comparison studies with respect to AChE inhibition showed that the potency of HupA was similar or superior to the inhibitors currently being used in AD treatment (Table 1)[11–13,19–21]. Based on the 50% inhibitory concentration (IC50), HupA was more potent than tacrine, physostigmine, galanthamine, and rivastigmine with respect to inhibition of AChE activity, whereas HupA was the least potent BuChE inhibitor among the inhibitors tested[11,21–23]. The IC50 ratio of HupA for BuChE:AChE was much greater than those of the other 4 inhibitors. HupA exerted inhibitory effects on AChE from different sources to a similar extent but, interestingly, was a weaker inhibitor of human serum BuChE relative to BuChE from other sources. Studies using 8 and 4 cholinesterase isoenzymes, which were extracted from mouse and dog plasma, respectively, showed that HupA had a more selective inhibition on AChE isoenzymes, but had little or no inhibition on BuChE[24]. The better reversible effect of (±)-HupA on AChE was also demonstrated in studies with porcine intrinsic cardiac neurons, which express both AChE and BuChE[25].
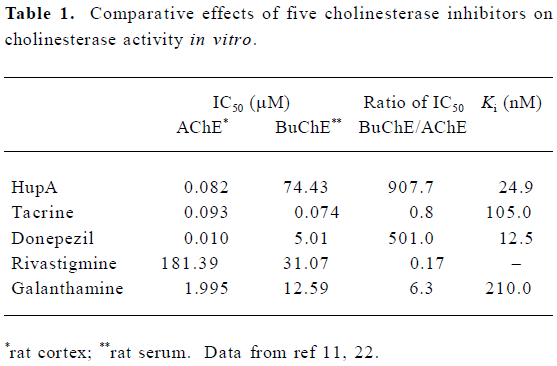
Full table
The apparent inhibition constants (Ki value) for AChE are in the nanomole range, which indicates that these inhibitors have high affinity for the enzyme. However, the doses of donepezil and tacrine used orally are much higher than that of HupA[26], which might be explained by their low bioavailability and/or by rapid metabolism.
AChE exists in multiple molecular forms that can be distinguished by their subunit associations and hydrodynamic properties[27,28]. In mammalian brain, the bulk of AChE occurs as a tetrameric, G4 form (10S) together with much smaller amounts of a monomeric, G1 (4S) form[29,30]. There is evidence that AChE inhibitors do not inhibit all forms equally well. Studies from our laboratory showed that HupA preferentially inhibited tetrameric AChE (G4 form), whereas tacrine and rivastigmine preferentially inhibited monomeric AChE (G1 form). Donepezil showed pronounced selectivity for G1 AChE in striatum and hippocampus, but not in cortex. Physostigmine showed no form-selectivity in any brain region. In cortex, the most potent inhibitors of G4 AChE were HupA and donepezil. The potent inhibitors of cortical G1 AChE were donepezil and tacrine. In hippocampus, HupA and physostigmine were the most potent inhibitors of G4 AChE, whereas donepezil and tacrine were the most potent against G1 AChE. In striatum, HupA and donepezil were the most potent against G4 AChE, and again donepezil was the most potent against G1 (Table 2). It is well known that approximately 60%–90% of G4 AChE is ectocellular, and is the major form for metabolizing ACh. The G4 AChE is the physiologically relevant form at cholinergic synapses, and its inhibition would be expected to prolong the action of ACh. The results mentioned above suggest that the use of AChE inhibitors in the treatment of AD must consider both form-specific and region-specific characteristics of AChE inhibition[12].
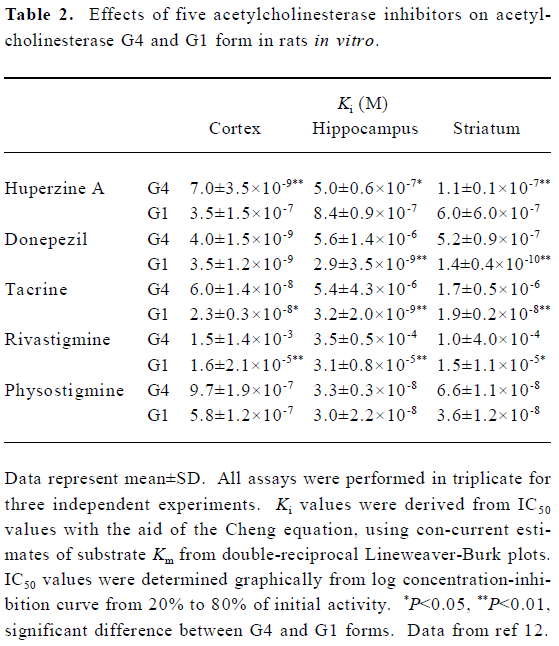
Full table
Significant inhibition of AChE activity was demonstrated in the cortex, hippocampus, striatum, medial septum, medulla oblongata, cerebellum, and hypothalamus of rats that were killed 30 min following the administration of HupA at several dose levels compared with the saline control[26,31,32]. There was a clearly dose-dependent inhibition of AChE in the brain region by HupA. In contrast to the inhibition of AChE activity in vitro, the relative inhibitory potency of oral HupA on cortex AChE was found to be approximately 24- and 180-fold, on an equimolar basis, that of donepezil and tacrine, respectively[19]. Correlated to the dosage of AChE inhibition, however, only donepezil and tacrine produced significant BuChE inhibition in serum[19]. Tacrine was a more potent inhibitor of serum BuChE than that of brain AChE. The inhibitory potency of HupA on AChE differs from that of donepezil and tacrine following different routes of administration. HupA exerted an almost similar anti-ChE efficacy in rats following oral and ip administration, whereas tacrine ip produced a greater inhibition both on brain AChE and serum BuChE. At doses of 0.03 µmol/L (8 µg) and 0.06 μmol/L (16 µg), HupA significantly inhibited brain AChE activity 30 min after intraventricular injection, which was less potent than donepezil, but more potent than tacrine[26]. These findings indicate that HupA, in contrast to donepezil and tacrine, has higher oral bioavailability and better penetrability through the blood-brain barrier.
AChE inhibition in rat whole brain reached a maximum at 60 min and was maintained for 360 min following oral admini-stration of HupA, at a dose of 1.5 µmol/kg (3.6 mg/kg). Peak inhibition in cortex and serum was observed at 30–60 min, and inhibition exceeding 10% in the cortex was maintained between 15–240 min. The BuChE activity recovered to the control level at 360 min after administration of HupA, whereas 20% and 46% inhibition still existed for donepezil and tacrine, respectively[19]. The rapid decrease of AChE and BuChE activity seen in red blood cells and plasma, respectively, with HupA correlates with the short-lasting and mainly peripheral side effects[31]. Repeated oral doses (once daily for 8 d and 30 d) of HupA produced no significant difference in AChE inhibition as compared to a single dose, indicating that no tolerance to HupA developed[19,33].
The enantiomers of HupA differ greatly in their ability to inhibit AChE. At equivalent doses, (+)-HupA was much weaker than (–)-HupA in inhibiting AChE in NG108-15 cells[34]. Careful measurement of AChE inhibition in cell-free systems revealed that (–)-HupA was 49-fold more potent than (+)-HupA as shown both by IC50 values and Ki (Table 3). The natural isomer was also more selective for AChE, as shown by the 9-fold higher ratio of BuChE IC50 versus AChE IC50. Similar differences in anti-cholinesterase potency were seen when changes in ChE activity in whole brain, cortex and serum were compared 30 min after oral administration of (–)-HupA and (+)-HupA. Although both enantiomers of HupA produced a dose-dependent inhibition of ChE, (–)-HupA was approximately 50-fold more potent than (+)-HupA[34]. This difference in activity measured between the 2 enantiomers may be partially ascribed to the fact that the H-bond between the ethylidene methyl of (–)-HupA and His440 is absent in the (+)-HupA complex[35,36].
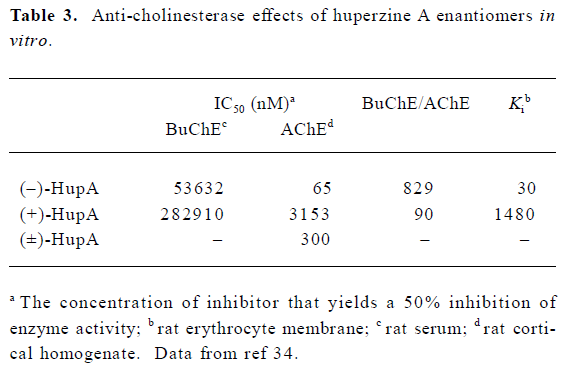
Full table
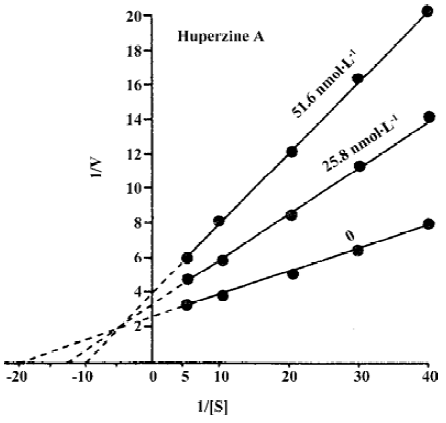
The mechanisms by which HupA inhibits AChE have been extensively studied by using kinetics[11,23,37], computer-aided docking studies[36,38] and X-ray crystallography approaches[35]. The Lineweaver-Burke plot representation of the inhibition of rat erythrocyte membrane AChE by HupA indicates a mixed competitive type of inhibition, because the intersection of the lines occurs in the second quadrant (Figure 2)[11,22,23]. A similar type of inhibition was found in porcine brain immobilized AChE[23]. Rat erythrocyte membrane AChE activity did not exhibit progressive decrease with prolonged incubation with HupA in vitro, and the AChE activity recovered to 94% of the control after being washed 5 times, indicating that the inhibitory action of HupA was reversible and different from that of isoflurophate (DFP)[11,23].
Immense effort has been directed towards gaining insights into the binding between HupA and AChE since 1991, when the 3-D structure of the native TcAChE was determined by using both X-ray crystallography and molecular modeling. The 2.5 Å resolved crystal structure of a Torpedo AChE-HupA complex demonstrated the “ingenious design” of the natural alkaloid[39] to bind more tightly and specifically to the enzyme than do other known AChE inhibitors such as tacrine and edrophonium. Furthermore, the refined structure clearly identifies the principal protein-ligand interactions responsible for the efficacy of the inhibitor upon binding to AChE[35]. The principal interactions include: (i) direct and strong hydrogen bonds between the carbonyl group of HupA and the hydroxy oxygen of Tyr130 (located at the peripheral site of the enzyme), as well as between the thylidene methyl group and the main-chain oxygen of His440 (a modality of the catalytic triad); (ii) indirect hydrogen bonds, mediated by 1 or 2 water molecules, between HupA and residues of the enzyme that constitute the active center (eg the ring nitrogen of HupA is hydrogen-bonded to carboxylic oxygen of Glu199, whereas the -NH3+ group is bonded to hydroxyl oxygen of Tyr 121); (iii) the cation-π interaction induced upon binding between the -NH3+ group of HupA and the aromatic rings of Trp84 and Phe 330 at the choline site (it should be noted that other reversible AChE inhibitors such as tacrine and edrophonium bind to the same site)[40]; and (iv) a large number of hydrophobic interactions that are established between a carbon atom of HupA and the various oxygen, nitrogen, or carbon atoms of the amino acid residues comprising the enzyme.
Computer-assisted docking studies and the resolution of high-resolution crystal structure data for the AChE-HupA complex provide a valuable platform for the rationalization of the higher selectivity of the inhibitor, as well as the distinct thermodynamic stability of the complex. For example, HupA can form an extra hydrogen bond with Tyr337 within the choline site that exists only in the mammalian homologue of AChE, but not in Torpedo enzymes and BuChE[41,42]. This particular interaction may be largely responsible for the much stronger inhibitory effect of HupA on mammalian AChE than that on the other 2 enzymes. In addition, the peptide flip between Gly117 and Gly118 (induced only by the binding of HupA) may explain why HupA possesses a longer residence time than other commonly used anticholinesterase agents[37].
Dvir et al reported that the oxyanion holes of AChE composed of Gly117, Gly118, and Gly119 were disrupted by HupA. The carbonyl oxygens of HupA appear to repel the carbonyl oxygen of Gly117, thus causing the peptide bond between Gly117 and Gly118 to undergo a peptide flip[36]. The new conformation is stabilized by Gly117O making H-bonds with Gly119N and Ala201N, the other 2 functional elements of the 3-pronged oxyanion hole characteristic of AChE[36]. It has been suggested that the peptide flip itself is responsible for the low on-rates observed for AChE inhibition by (–)-HupA[35]. Its stabilization may contribute to the low rates of dissociation observed[37].
Effects on cholinergic parameters
To study the effect of HupA on cholinergic transmission at mouse neuromuscular junctions in vitro, isolated mouse phrenic nerve-hemidiaphragm preparations were used with a conventional intracellular recording technique. HupA at a concentration of 1 µmol/L increased the amplitude, time-to-peak, and half-life of miniature end-plate potentials (MEPP) of muscle fiber[43]. HupA had no effect on resting membrane potentials, the mean quantal content of end-plate potentials and the frequency of MEPP of muscle fiber, indicating that the effects of HupA may not be mediated through presynaptic or postsynaptic mechanisms. In contrast to donepezil and tacrine, neither the appearance of giant MEPP nor slow MEPP was changed by HupA, ruling out the possibility of non-specific promoting effects on terminal ACh release[44]. In a study of toad paravertebral ganglia (PVG) using intracellular recording techniques[45], HupA, at concentrations of 0.3, or 1 µmol/L, did not change membrane potential or input resistance, but increased the rate of orthodromic action potential evoked by preganglionic stimulation, in contrast to physostigmine[46] and tacrine[47]. HupA increased exogenous ACh- but not carbachol-induced depolarization, indicating that the facilitating effect of HupA on ACh transmission is mainly mediated by its anti-AChE activity.
Neuronal nicotinic acetylcholine receptors (nAChR) are involved in cognition and may play a role in AD. Studies on nAChR in rat hippocampal CA1 interneurons in slices using patch-clamp techniques showed that HupA had no significant effect on either the amplitude or kinetics of α7 nAChR activated by ACh, but slowed the rate of recovery from desensitization through an indirect mechanism. For non- α7 receptors, HupA significantly increased the amplitude and decay phase for responses induced by ACh (but not carbachol), also through an indirect mechanism. The results suggested that AChEI were likely to be important regulators of cholinergic signaling in the hippocampus[48].
In isolated rat phrenic nerve-diaphragm preparation, HupA significantly increased the amplitude of muscle contraction induced by stimulating the nerve. The anticurare effect of HupA was found to be much more potent than that of neostigmine in anesthetized rat sciatic-tibialis preparation. The salivation induced by HupA was less potent than that induced by neostigmine[49].
HupA produced an altered electroencephalography (EEG) result in conscious rabbits, which showed a decrease in lower frequency components and the total EEG power in the cortical area, and the dominant frequency changed from delta rhythm to theta rhythm in the hippocampus. These effects are cholinergic in nature and can be reversed by scopolamine or atropine[49–51].
The acetylcholine potentiating action of HupA has been observed in the frog rectus abdominus muscle, rat phrenic nerve diaphragm preparation, guinea pig ileum, and human iris sphincter muscle. HupA has greater acetylcholine potentiating activity on vertebrate muscles than does physostigmine[49,52].
In the hippocampus, high-affinity choline transport was reduced by 28% after multiple ip doses of 0.5 mg/kg HupA[33]. Because the effect of HupA was completely reversible with time and not mediated through a direct interaction with the transporter, this effect was probably mediated through regulatory control of high-affinity choline transport in response to ACh increases following ChE inhibition rather than by directly acting on the transporter.
Studies on the displacement of [3H]QNB- and [3H](–)nicotine-specific binding showed that HupA had little direct effect on cholinergic receptors compared with tacrine and heptylphysostigmine[31,53]. The concentration required to display 20% specific binding was 20 µmol/L for [3H](–)nicotine and 160 µmol/L for [3H]QNB, indicating that lower concentrations of HupA have a stronger displacing effect on [3H](–)nicotine- than on [3H]QNB-specific binding. A stronger effect of a low dose HupA on central nicotinic receptors may constitute an additional therapeutic advantage in the treatment of AD. The low level of ACh synthesis in the cortex of a patient with AD may maintain presynaptic nicotinic receptors in an active state with no desensitization[54]. In such a state, nicotinic receptors may become more sensitive to stimulation by HupA.
HupA at concentrations of 1–100 µmol/L tested in vitro did not affect the electrically evoked release of [3H]ACh from rat cortical slices[31], which contrasted with the decreased release seen with tacrine, physostigmine, and metrifonate[55]. This finding suggests that HupA might not have exerted direct action on the cholinergic presynaptic receptors controlling ACh release.
Effects on brain neurotransmitters
Compared with other inhibitors being used in the therapy of AD, HupA produced a more prolonged increase of ACh levels than did tacrine, donepezil, rivastigmine, physostig-mine, or metrifonate after systemic administration[13,31,32,53,56]. There is considerable regional variation in the increase of ACh levels after HupA administration, with maximal increase seen in frontal and parietal cortex, and smaller increases in the striatum and cerebellum[31]. Considering that ACh level is particularly low in the cerebral cortex of patients with AD[57], this particular regional specificity produced by HupA may constitute a therapeutic advantage. A positive correlation was seen between ACh levels and AChE activity in the frontal cortex and whole brain[13,31,32,53]. Studies using microdialy-sis techniques in conscious, freely moving rats showed that HupA dose-dependently elevated the level of ACh in cortex and hippocampus. The time course of cortical AChE inhibition with HupA mirrored the increase of ACh at the same doses[13]. This result is consistent with the idea that the increase in extracellular ACh was due primarily to the inhibition of cortical AChE. In molar terms, HupA was 8- and 2-fold more potent than donepezil and rivastigmine, respectively, in increasing cortical ACh levels, with a longer-lasting effect (Figure 3)[13]. Tolerance or accumulation of increasing ACh level was not formed after multiple administration of HupA (Liang YQ et al, unpublished data). HupA did not alter choline levels[32] or the activity of choline acetyltransferase (ChAT) in any region of the rat brain assayed[32,33], suggesting that the increase of ACh levels by HupA was not likely to be mediated through an increase in the rate of ACh synthesis.
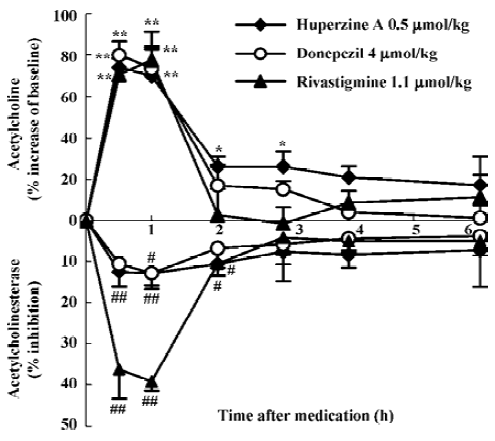
Brain norepinephrine (NE) and dopamine (DA) levels increased significantly following either systemic administration of HupA or local administration of HupA through microdialysis, but 5-HT level was not affected[56]. HupA produced an 11- and 2-fold more potent increase in the DA level of the middle prefrontal cortex than donepezil and rivastigmine, respectively. The increasing effect of HupA on DA level was more potent than that on NE level (Liang YQ et al, unpublished data). Oxotremorine (a muscarinic agonist) and mecamylamine (a nicotinic antagonist) completely blocked the increasing effects of HupA on DA and NE levels, suggesting that ACh regulation by presynaptic ACh muscarinic receptors or nicotinic receptors accounts for the effect of HupA on DA and NE. These effects may be involved in the memory improvement effected by HupA (Liang YQ et al, unpublished data).
Enhancing effects on cognition
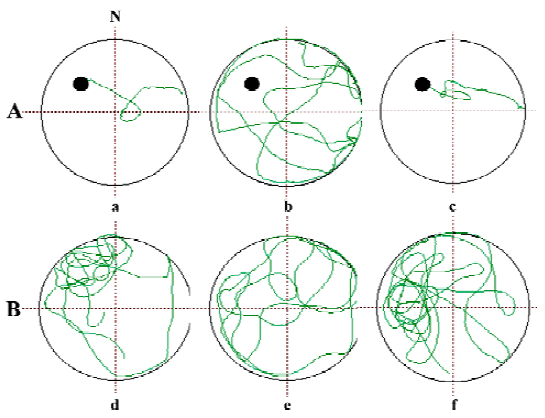
HupA has been found to be an effective cognition enhancer in a number of different animal species. Enhancement of learning and memory performance was demonstrated in passive footshock avoidance[58–61], water maze escape task[63,64], and spatial discrimination in a radial arm maze[10,65], as well as in delayed response performance in monkeys[66,67]. Beneficial effects were seen not only in intact adult rodents, aged rodents[58,64] and monkeys[66], but also in rodents cognitively impaired by scopolamine[10,31,50,60–63,68], AF64A[26,69], electroshock[61,68], cycloheximide[61], NaNO2[61], CO2[58,60], and D-galactose[70]. Inverted U-shaped dose-response curves typical of cognition enhancers were found with HupA. The duration of improvement induced by oral HupA on learning and memory retention processes were longer than those induced by physostigmine, galanthamine, and tacrine, respectively[71]. HupA has a higher efficacy than tacrine and donepezil given orally[19]. The improvement effected by HupA was more pronounced in working memory than in reference memory[10], which may benefit AD patients because the cognitive deficits in memory of recent events are more severe in AD. In aged rats, HupA could significantly reduce the latent period of finding the platform and increased the time in the probe quadrant in the Morris water maze performance[64]. In addition, HupA improved cognition in cholinergically lesioned rats[58,61] and the spatial working memory deficit induced by lesions in the nucleus basalis magnocellu-laris[72].
HupA reversed memory deficits induced by bilateral injection of scopolamine and muscimol (a GABAA agonist) into hyperstriatum ventrale in chicks. The improvement was observed from 30 min to 90 min, but not at 10 min after training, indicating that HupA participated in the modulation of intermediate-term memory and long-term memory formation in a passive avoidance task. This finding suggests that HupA improved memory formation not only by acting as a highly potent inhibitor of AChE, but also by antagonizing the effects mediated through the GABAA receptor[62]. It is well known that deficiencies in ACh and GABA content have been observed in the cortical regions of AD brains[73]. Anatomical evidence also suggests an important interaction between GABA and cholinergic neurons in the septum and hippocampus[74]. Thus, a drug that is able to enhance synaptic ACh and also antagonize the GABAA receptor could be ideal for treatment of AD.
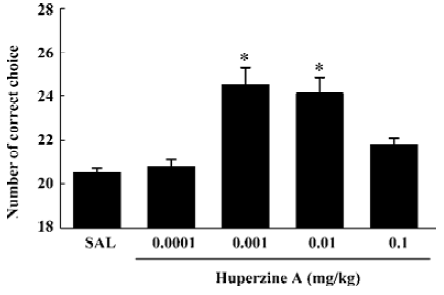
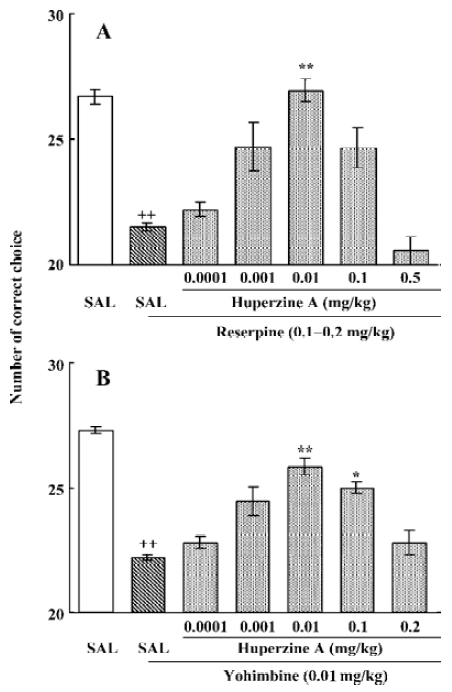
To extend the antiamnesic effect of HupA to non-human primates, HupA was evaluated for its ability to reverse the deficits in spatial memory produced by scopolamine in young adult rhesus monkeys or those occurring naturally in aged monkeys using a delayed-response task[66]. HupA (0.01–0.1 mg/kg, im) improved the memory deficits induced by scopolamine in young adult monkeys. In aged monkeys, HupA (0.001–0.01 mg/kg, im) significantly increased choice accuracy in delayed response performance (Figure 4). The beneficial effects of HupA were long lasting. Monkeys retained improved performance for approximately 24 h after a single injection of HupA. Given that the NE and DA levels decreased significantly with aging in monkeys[75], the effect of HupA on memory may be associated with increased levels of NE and DA. HupA (0.01–0.1 mg/kg, im) also produced significant improvement on reserpine- (a catecholamine depleting agent) and yohimbine- (an α2-adrenoceptor antagonist) induced memory impairments in monkeys (Figure 5). These effects are primarily due to the increase of NE level by HupA, which might stimulate the α2-adrenoceptor in the prefrontal cortex to improve the delayed response performance[67].
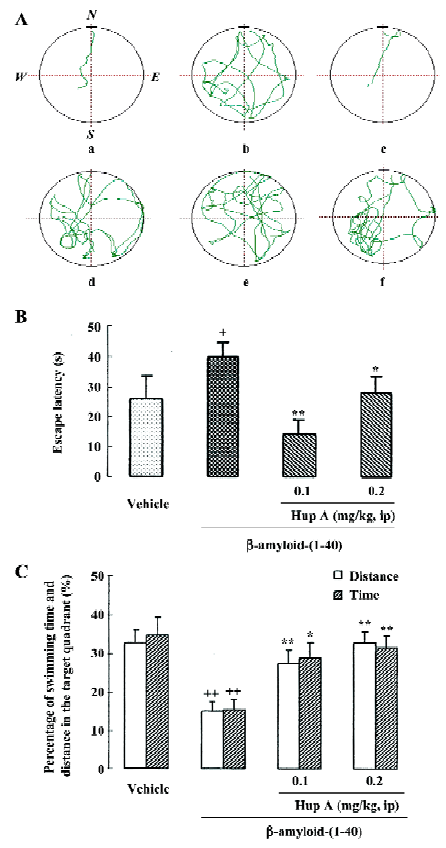
Deposition of β-amyloid protein is considered a crucial event in initiating the neuritic and neuronal degeneration in AD, which mainly affects the areas involved in cognitive function, such as cortices, some limbic structures, and the forebrain nuclei projecting to those areas. Repeated icv infusion of β-amyloid protein-(1-40) induced marked amnesic effects along with signs of neurodegeneration and extracellular amyloid deposits throughout the frontoparietal cortex and hippocampus, which indicated that β-amyloid protein deposition in the brain was related to cognitive impairment, hypofunction of cholinergic neurons and neuronal death. Daily intraperitoneal administration of HupA for 12 consecutive days produced significant reversals of the β-amyloid-induced deficit in learning a water maze task (Figure 6). Treatment with HupA also attenuated the neuronal degeneration induced by Aβ1-40 in the cortex and hippocampus, indicating that neuroprotection is involved to some extent in the favorable effect of HupA on Aβ-induced memory deficits[76].
It is well-documented that N-methyl-D-aspartate (NMDA)-receptor activation mediates the generation of long-term potentiation (LTP), a cellular process that underlies learning and memory[77,78]. There is evidence that the suppressive action of Aβ on LTP in both CA1 and dentate gyrus operates via an NMDA receptor-independent pathway that involves cholinergic terminals in the hippocampus. It is of some interest that HupA (1.0 µmol/L) was found to enhance LTP, but at a much lower dose (0.1 µmol/L) largely blocked the suppressive effects of Aβ on LTP induction[79,80], which might involve the mechanism of HupA against Aβ-induced cognitive deficits.
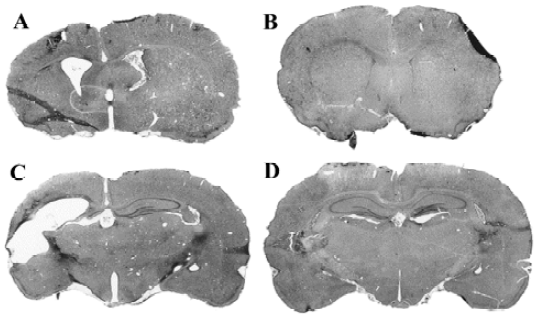
Apart from AD, the most common dementia in the elderly is vascular dementia (VD). This disorder, like AD, presents as a clinical syndrome of intellectual decline produced by ischemia, hypoxia, or hemorrhagic brain lesion. Rats with permanent bilateral ligation of the common carotid arteries exhibit learning and memory impairments and neuronal damage resembling those in VD. In these rats, daily oral administration of HupA produced a significant improvement of the deficits in learning the water maze task, beginning 28 d after ischemia, along with approximately 33%–40% inhibition of AChE activity in the cortex and hippocampus[81]. Similar cognitive improvement of HupA was observed in a gerbil model of transient global ischemia[82]. The protective effects of HupA against hypoxic-ischemic (HI) brain injury were also found in neonatal rats. Unilateral HI brain injury was produced by ligation of the left common carotid artery followed by 1 h hypoxia with 7.7% oxygen in 7-d-old rat pups. After 5 weeks, HI brain injury in rat pups resulted in working memory impairments as shown by increased escape latency in a water maze and reduced time spent in the target quadrant. Rats treated with HupA at doses of 0.05 or 0.1 mg/kg ip for 5 weeks after HI injury performed better than saline-treated HI rats, and neuronal damage in the ipsilateral hemisphere was attenuated (Figure 7). These findings suggest that HupA might be beneficial in the treatment of HI encephalopathy in adults and neonates[83].
Neuroprotective effects
Several neurodegenerative disorders such as AD, cerebral ischemia-reperfusion injuries and head injuries are thought to be related to changes in oxidative metabolism. Increased oxidative stress, resulting from free radical damage to cellular function, can be involved in the events leading to AD, and is also connected with lesions called tangles and plaques. Plaques are caused by the deposition of Aβ and are observed in the brains of AD patients[2,84–86]. Studies show that oxygen radicals initiate amyloid build-up, leading to neurodegeneration[85,87]. HupA has been found to protect against H2O2- and Aβ-induced cell lesion, decrease the level of lipid peroxidation, and increase antioxidant enzyme activities in rat PC12 and NG108-15 cell lines and primary cultured cortical neurons (Figure 8)[34,88–91]. Following 6 h exposure of the cells to H2O2 (200 µmol/L) or 48 h exposure to Aβ25-35 (1 µmol/L), a marked reduction in cell survival, activity of glutathione peroxidase (GSH-Px) and catalase (CAT), as well as increased production of reactive oxygen species (ROS) and malondial-dehyde (MDA) were observed. Pretreatment of the cells with HupA (0.1–10 µmol/L) 2 h before H2O2 or Aβ exposure significantly elevated cell survival. HupA reversed H2O2- and Aβ-induced decreases in GSH-Px and CAT activity, as well as causing increases in the production of ROS, MDA and superoxide dismutase (SOD).
Oxygen-glucose deprivation (OGD) for 30 min caused death in more than 50% of rat pheochromocytoma PC12 cells, along with major changes in morphology and biochemistry, including elevated levels of lipid peroxide, SOD activity and lactate concentration. Cells pretreated for 2 h with HupA (0.1, 1, or 10 µmol/L), however, had increased survival and reduced biochemical and morphologic signs of toxicity. HupA protected PC12 cells against OGD-induced toxicity most likely by alleviating disturbances of oxidative and energy metabolism[92].
In rat studies, intracerebroventricular infusion of β-amyloid1-40 (800 pmol×3) induced significant morphological injury and decreases in cortical ChAT activity. Daily ip administration of HupA for 12 consecutive days attenuated the loss of ChAT activity in the cerebral cortex and the neuronal degeneration induced by β-amyloid1-40[76].
MDA level and manganese-SOD (Mn-SOD) activity in hippocampus, cerebral cortex, and serum of aged male rats were 2.3–2.8 times and 1.8–2.8 times greater, respectively, than those of adult male rats. HupA (0.05 mg/kg, ig) markedly lowered the levels of MDA and the activities of Mn-SOD in aged male rats following 7–14 consecutive days of daily administrations[93]. A reduction of oxygen free radicals in the plasma and erythrocytes was also demonstrated in a clinical study[16].
In chronic cerebral hypoperfused rats, HupA restored the decrease in ChAT activity in the hippocampus, improved neuronal morphological damage, and restored SOD and lipid peroxides activities, as well as lactate and glucose concentrations to their normal levels[81]. Similar protective effects of HupA were observed in the studies of transient global ischemia in gerbils[82]. The protective effect of HupA on HI brain injury has also been found in neonatal rats[81]. A unilateral HI brain injury was produced by the ligation of the left common carotid artery (CCA) followed by 1 h hypoxia with 7.7% oxygen in 7-d-old rat pups. After 5 weeks, the HI brain injury in rat pups caused damage in the ipsilateral striatum, cortex and hippocampus, as well as a marked reduction in CA1 neuron density. These neuropathologic signs were attenuated by the administration of HupA at a dose of 0.1 mg/kg (Figure 9). These results raise the possibility that HupA may be potentially useful in treating HI encephalopathy in neonates.
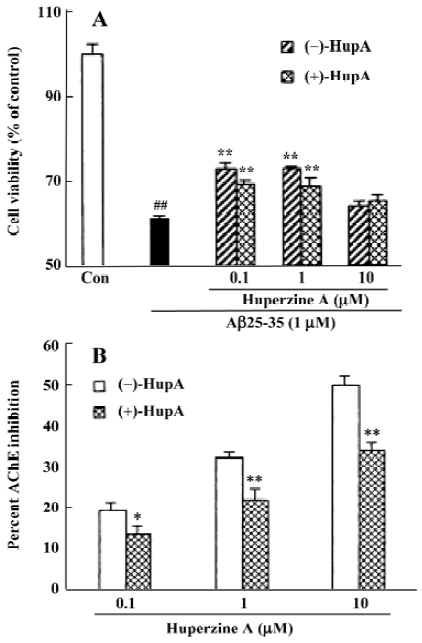
In studies carried out to examine the stereoselectivity of the cellular protective effect induced by (–)-HupA, it was found that (–) and (+) HupA exerted similar potency in protecting against the cellular toxicity induced by Aβ25-35. This result contrasted with the stereoselectivity of cholinesterase inhibition in vitro and in vivo (Figure 10)[34]. The ability of HupA to inhibit the catalyzing activity is not parallel to its neuroprotective effect, implying that the cytoprotective effect of the 2 enantiomeric forms of HupA might relate to the non-catalytic action of AChE.
It was recently reported that HupA exerted a neuroprotec-tive effect via modulating the intracellular Ca2+ ([Ca2+]i) level together with the mRNA transcription of calmodulin (CaM) and calmodulin-dependent protein kinase II (CaMPK II) in hippocampal neurons[94]. Mice given repeated CCA ligation-reperfusion treatment showed a marked increase in [Ca2+]i, and a decrease in CaM and CaMPK II mRNA levels in hippocampal neurons. Daily oral administration of HupA (0.05 mg/kg) for 30 consecutive days significantly reversed the shift in [Ca2+]i, CaM and CaMPK II mRNA levels induced by ischemia.
The findings mentioned above indicate that HupA has protective effects against free radical-, ischemia- and Aβ-induced cell toxicity, which might be beneficial in the treatment of patients with AD and VD.
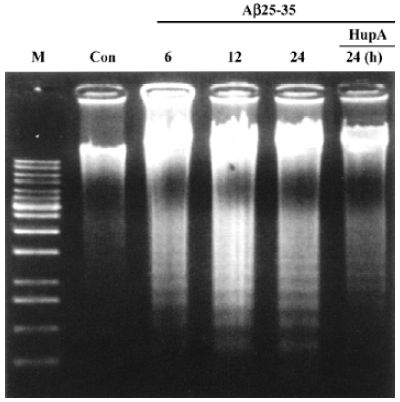
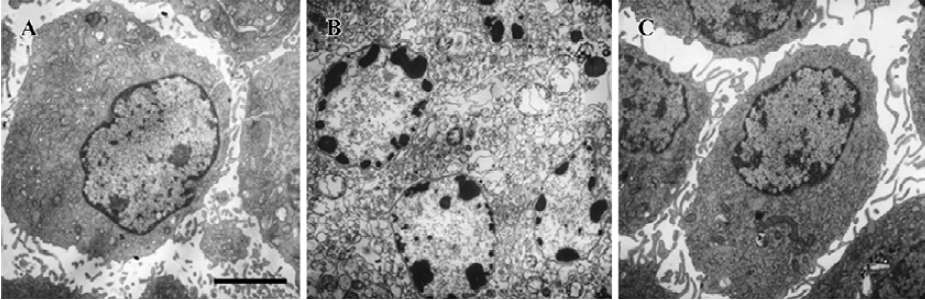
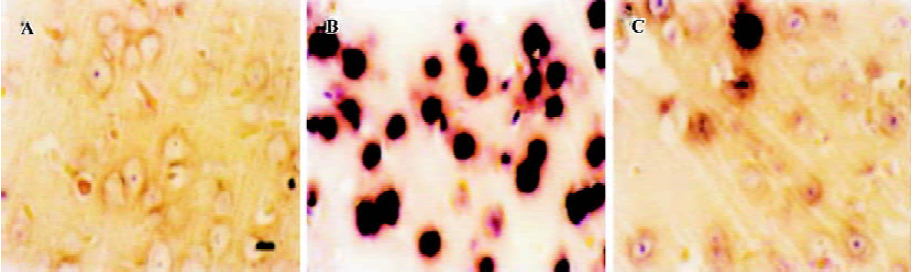
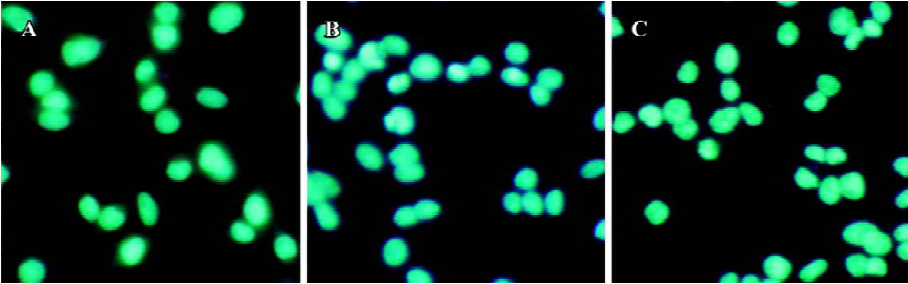
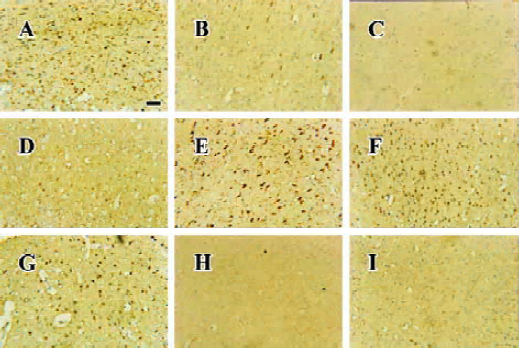
Apoptosis is the process by which neurons die during normal development and is also a feature of chronic and acute neurodegenerative diseases and stroke[95]. In accordance with previous reports, studies from our lab demonstrate typical apoptotic changes when neuron-like cells are exposed to stressors such as H2O2[96], Aβ peptides[91], oxygen-glucose deprivation (OGD)[97], serum deprivation[98], the protein kinase C (PKC) inhibitor staurosporine[99], and global ischemia[82]. These changes include DNA laddering (Figure 11), cell shrinkage, generation of nuclear apoptotic bodies, terminal deoxyribonucleo-tidyl transferase-mediated dUTP-digoxigenin nick end-labeling (TUNEL) positive staining (Figure 12), chromatin condensation (Figure 13), and other classic hallmarks of apoptosis (Figure 14)[91,96,97,99]. Such abnormalities are markedly relieved by HupA. Administration of HupA (0.1 or 0.2 mg/kg, ip, per day) for 12 consecutive days conferred substantial neuroprotection on rats that received icv injections of β-amyloid1-40 (800 pmol×3): the number of apoptotic-like neurons were markedly reduced[76]. In primary cultured neurons, preincubation with HupA at concentrations higher than 0.01 µmol/L led to a large dose-dependent attenuation of cell toxicity induced by Aβ25-35[91]. Moreover, HupA (1 µmol/L) caused large reductions in the amounts of subdiploid DNA detected in a flow cytometry assay and weakened the ladder pattern on agarose gel electrophoresis, which is typically seen after exposure to Aβ (Figure 11)[91]. The inhibition of ROS formation may involve the anti-apoptotic actions of HupA[91].
The cellular commitment to apoptosis is regulated by the Bcl-2 family of proteins. High levels of Bcl-2 expression will inhibit apoptosis. In contrast, an increased expression of P53 and Bax is associated with the initiation of apoptosis[100]. Treatment with HupA attenuated H2O2-, Aβ- and OGD-induced overexpression of mRNA and protein levels for c-jun, Bax and P53, and downregulated that of Bcl-2 to normal levels (Figure 15)[76,96,97].
In the mitochondrial-mediated cell death pathway, a key step is transient opening of the mitochondrial permeability transition (MPT), involving a non-specific increase in the permeability of the inner mitochondrial membrane[101,102]. In this process, cytochrome c moves from the intermembrane space into the cytoplasm[103], where it binds to Apaf-1 (the molecular core of apoptosome, which executes mitochondria-dependent apoptosis). In the presence of dATP, this complex polymerizes into an oligomer known as the apoptosome. The apoptosome activates the protease, caspase-9, which in turn activates caspase-3. The cascade of proteolytic reactions also activates DNase, which leads to cell death[104]. Our recent results showed that PC12 cells, when pre-incubated with HupA at concentrations above 0.01 µmol/L, were markedly protected against apoptosis induced by Aβ, with a significant reduction in mitochondrial swelling and improved mitochondrial membrane potentials (Gao X et al, unpublished data).
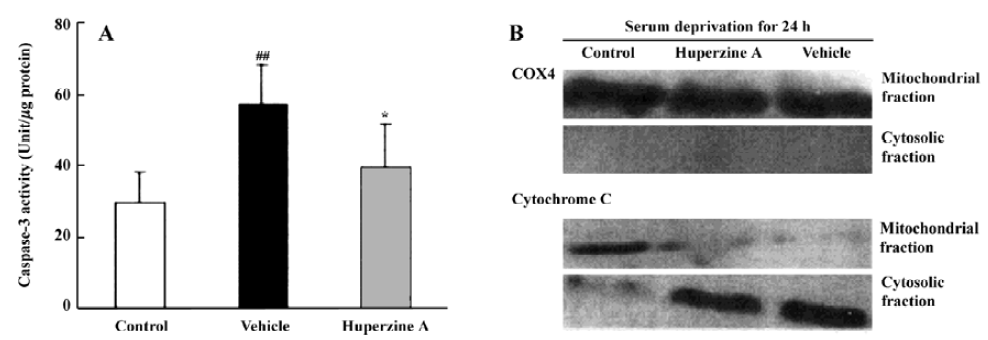
A series of studies were carried out in our laboratory that focused on caspase activation in primary cultures of rat cortical neurons subjected to a variety of stresses. Measurements of caspase-3-like fluorogenic cleavage demonstrated that HupA (1 µmol/L) attenuated the Aβ25-35-induced increase in caspase-3 activity at 6, 12, 24, and 48 h[91]. Western blot analyses confirmed these results at the protein level. HupA also inhibited caspase-3 activation in models of apoptosis induced by serum deprivation and staurosporine treatment. The apoptosis induced by serum deprivation for 24 h was accompanied by enhanced caspase-3 activity and a release of mitochondrial cytochrome c into the cytosol[98]. HupA (0.1–10 µmol/L) improved neurons survival, inhibiting the rise in caspase-3 activity and protein expression (Figure 16)[98]. Likewise, cell survival was greatly enhanced when HupA (0.1–100 µmol/L) was introduced 2 h before 24 h exposure to 0.5 µmol/L staurosporine. Staurosporine-induced DNA fragmentation, upregulation of the pro-apoptotic gene bax, downregulation of the antiapoptotic gene bcl-2, and decrease in caspase-3 proenzyme protein level were all attenuated by HupA at dose of 1 µmol/L[99].
A potassium channel with delayed rectifier characteristics may play an important role in Aβ-mediated toxicity[105]. The upregulation of an outward K+ current known as Ik mediates several forms of neuronal apoptosis and might specifically contribute to the pathogenesis of Aβ-induced neuronal death. Exposure to 20 µmol/L Aβ25-35 or Aβ1-42 is known to enhance the apoptosis-related current, IK[106]. Expression of wild-type PS-1 or PS-2 increases outward K+ current densities in HEK293 cells relative to untransfected or mock-transfected cells[107]. These data raise the intriguing possibility that manipulations aimed at reducing outward K+ current may provide an approach to reducing neuronal degeneration in patients with AD.
HupA reversibly inhibited the fast transient potassium current (IA) in CA1 pyramidal neurons acutely dissociated from rat hippocampus. The effect was voltage-independent and insensitive to atropine. HupA slowed down the decay of IA and its recovery from inactivation and showed no effect on steady-state inactivation, but hyperpolarized the activation curve of IA by 6 mV, suggesting that HupA may act as a blocker at the external mouth of the A channel[108]. In addition, HupA inhibited another important outward K+ current, the sustained potassium current (IK) in a voltage-dependent manner in acutely dissociated rat hippocampal neurons. The effect was insensitive to atropine. HupA hyperpolarized the activation curve of the current by 16 mV, and markedly prolonged the decay time constant τ2[109]. Because outward K+ current has proven very important in apoptosis induction, the reversible inhibitory effects of HupA on IA and IK might contribute to its anti-apoptotic effect.
In light of these findings, we can conclude that HupA, in addition to being a potent, highly specific and reversible inhibitor of AChE, possesses the ability to protect neurons against cytotoxicity and apoptosis induced by H2O2, Aβ, OGD, ischemia, serum deprivation and staurosporine. The protective and anti-apoptotic actions of HupA may involve inhibiting the production or the effects of ROS, improving energy metabolism, regulating apoptosis-related gene expression, protecting mitochondrial function, as well as modulating intracellular Ca2+ concentrations and inhibiting outward K+ currents. The neuroprotective effect of HupA is not correlated with its AChE inhibitory activity. These findings suggest that the therapeutic effects of HupA may be exerted via a multi-target mechanism.
Protection of HupA against glutamate-induced cytotoxicity
Glutamate is the main excitatory neurotransmitter in the central nervous system (CNS), with important roles in neurotransmission and functional plasticity. Excitatory amino acid neurotransmitters are also involved in CNS pathology. The deleterious effects of overstimulation with excitatory amino acids have been implicated in a variety of acute and chronic neurodegenerative disorders, including ischemic brain damage, AD and neuronal cell death[110–115]. Glutamate-mediated overactivation of receptors induces excessive Ca2+ influx, which results in elevated intracellular Ca2+ concentrations[116,117] with serious consequences such as necrosis and apoptosis[118]. Blockade of glutamate receptors prevents most of the Ca2+ influx and neuronal cell death induced by glutamate exposure[119,120].
It has been reported that HupA protects against gluta-mate-induced toxicity. HupA (100 µmol/L) was found to decrease neuronal cell death caused by a toxic level of glutamate. In those experiments, HupA reduced glutamate-induced calcium mobilization but did not affect the increase in intracellular free calcium induced by exposure to high KCl or a calcium activator Bay-K-8644[121]. HupA dose-dependently inhibited the NMDA-induced toxicity in primary neuronal cells, most likely by blocking NMDA ion channels and inhibiting the subsequent Ca2+ mobilization at or near the phencyclidine (PCP) and MK-801 ligand sites[122]. HupA reversibly inhibited NMDA-induced current in acutely dissociated rat hippocampal pyramidal neurons and blocked specific [3H]MK-801 binding in synaptic membranes from rat cerebral cortex[123]. Of all AChE inhibitors tested, HupA is the most powerful both in protecting mature neurons and in blocking the binding of [3H]MK-801. The effect was non-competitive, and showed neither “voltage-dependency” nor “use-dependency”[124]. HupA acts as a non-competitive antagonist of the NMDA receptors, via a competitive interaction with one of the polyamine binding sites[125]. It is interesting that natural (–)-HupA and synthetic (+)-HupA reduced the binding of [3H]MK-801 with similar potency[126], indicating that HupA inhibits NMDA receptors in rat cerebral cortex without stereoselectivity. This result is in dramatic contrast with the stereoselective inhibition of acetylcholinesterase.
Neuronal cell death caused by overstimulation of gluta-mate receptors has been proposed as the final common pathway for a variety of neurodegenerative diseases, including AD. The ability of HupA to attenuate glutamate-mediated neurotoxicity may be one additional reason for considering this agent as a potential therapeutic for dementia and as a means of slowing or halting the pathogenesis of AD at an early stage[122].
Effects on secretory amyloid precursor protein and protein kinase C-α
Aβ is a self-aggregating 39–43-amino-acid peptide that originates from a larger polypeptide that is known as the Alzheimer’s amyloid precursor protein (APP). Alternate pathways for APP processing have been described: the non-amyloidogenic secretory pathway, which releases a soluble ectodomain (APPs) and prevents Aβ formation[127], and the endosomal-lysosomal pathway, which produces amyloido-genic products[128]. The amyloid hypothesis of AD[129,130], which is focused on the potential toxic role of an excessive production of Aβ, suggests that the aberrant metabolism of APP is a central pathogenetic mechanism for the disease.
Several factors can affect the secretory non-amyloido-genic pathway of APP. For example, the stimulation of phospholipase C (PLC)-coupled receptors, such as muscarinic m1 and m3, has been shown to potentiate the secretion of APPs in cell cultures. These effects are probably mediated mainly by PKC[131]. It has also been reported that several AChEI affect APP processing in addition to the catalytic function of AChE[6,132,133].
HupA can alter the disturbance of PKC and APPs induced by Aβ in both rats and the HEK293sw cell line[134]. The levels of APPs and PKCα were significantly decreased after infusion with Aβ1-40 in rats. HupA caused a marked reduction of these changes (Figure 17), but had no significant effects on APPs or PKCα in normal rats. In HEK293sw cells, HupA increased the levels of APPs and PKCα, but had no effect on the levels of PKCδ and PKCε. These findings suggest that HupA may reduce APP processing through upregulating PKC, especially PKCα levels.
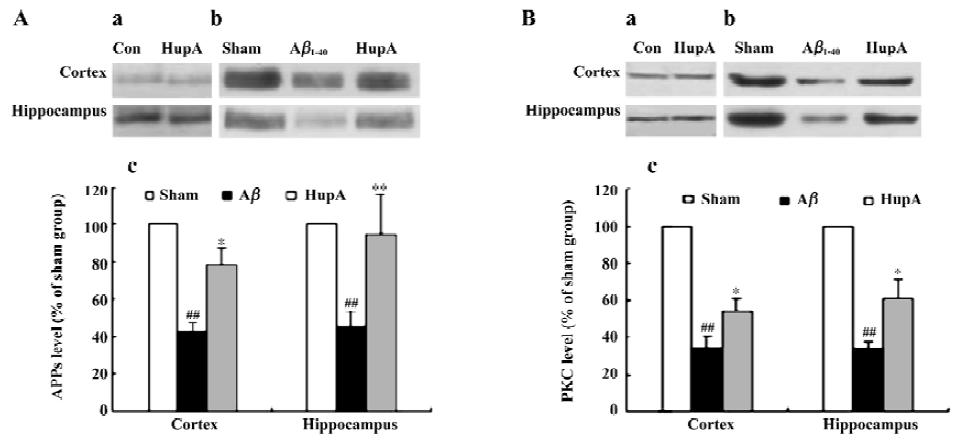
In an attempt to clarify the receptor mechanisms involved in such effects, we found that HEK293wt cells treated with scopolamine, a nonselective muscarinic antagonist, partly blocked the HupA-induced increase in the levels of APPs and PKC. In contrast, the nicotinic antagonist mecamylamine had no effect, suggesting that stimulation of the non-amyloidogenic secretory pathway of APPs metabolism by AChE inhibition is probably accomplished via an effect on the muscarinic receptors/PKC cascade (Yan H et al, unpublished data). Because PKC is a key enzyme in signal transduc-tion, and because APPs itself has neuroprotective effects, modulating the levels of these 2 proteins by HupA may be beneficial in AD therapy.
Effects on nerve growth factor
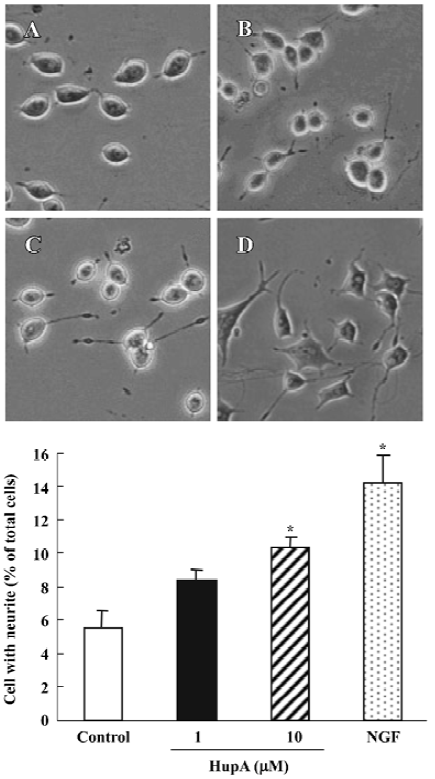
Nerve growth factor (NGF) is a member of the neurotrophin family that promotes the survival and outgrowth of central cholinergic neurons[135]. The decrease in trophic support for the neurons in the aging brain is associated with neuronal death and appearance of neurodegenerative disorders such as AD[136]. Accumulated data suggest that cholinergic mechanisms are involved in the regulation of NGF synthesis and release, and some AChE inhibitors are known to exert NGF-like activities by potentiating the neuritogenic effect of NGF[137]. HupA has been shown to increase neurite outgrowth from undifferentiated PC12 cells (Figure 18) and to enhance the expression and secretion of NGF, as well as increase p75NTR mRNA in primary astrocytes (Figure 19). These effects suggest the possibility that HupA increases the NGF-induced enhancement of neuron survival and function improvement, which is helpful in the rescue of injured neurons in neuro-degenerative disease. In addition to inhibiting AChE activity, AChE mRNA expression and protein levels were significantly upregulated after treatment with HupA. Accumulating evidence indicates that AChE may influence neurite outgrowth through a non-catalytic mechanism[138]. Hence, the effect of HupA on neurite outgrowth may be associated with the level of AChE expression[139].
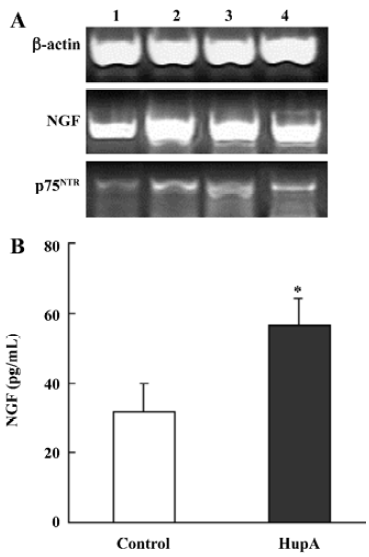
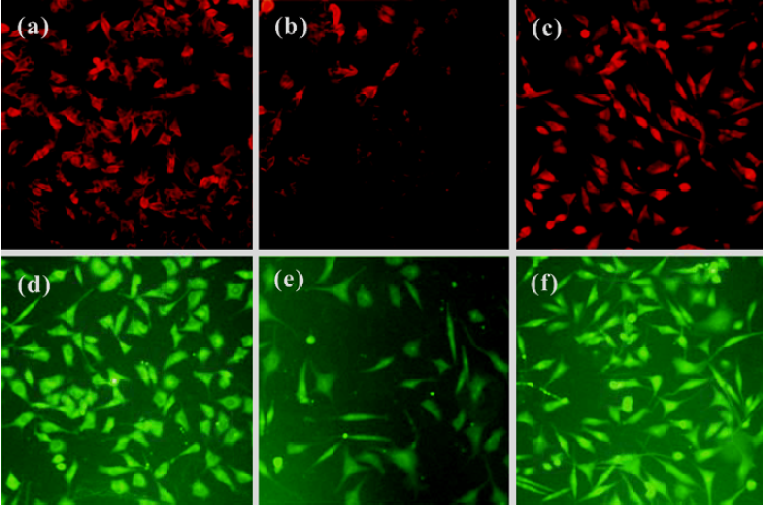
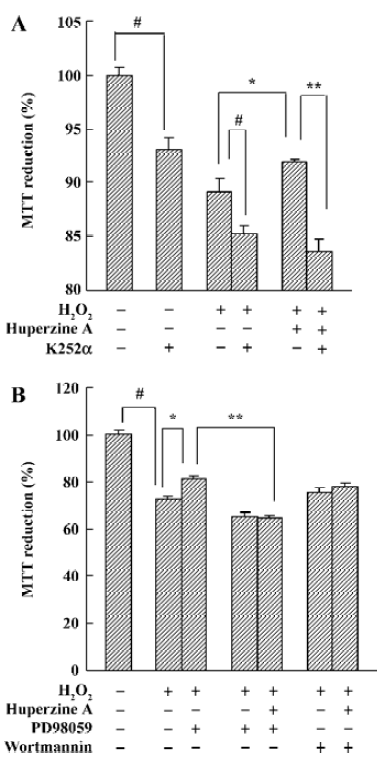
Neurotrophic factors such as NGF not only promote the survival of responsive neurons but also protect them from oxidative injury. Our study with SH-SY5Ycells used H2O2 to generate an oxidative stress sufficient to cause cell loss along with a substantial decrease in the mRNA and protein levels of NGF, neurotrophin receptor p75 (p75NTR) and tyrosine kinase A (TrkA). HupA not only reduced the overt signs of cytotoxicity, but also preserved the expression of NGF and its receptors (Figure 20). These neuroprotective effects of HupA on H2O2-induced cytotoxicity were blocked by the TrkA phosphorylation inhibitor K252a, and were antagonized by the mitogen-activated protein (MAP) /extracellular signal-regulated kinase (ERK) inhibitor PD98059 (Figure 21). These findings indicate that the NGF and TrkA receptor mediate key events required for the neuroprotective actions of HupA. Among the downstream signaling events triggered by the action of NGF at the TrkA receptor, activation of the MAP/ERK kinase pathway may be particularly important for the ability of HupA to protect SH-SY5Y cells against oxidative stress[140].
Pharmacokinetics
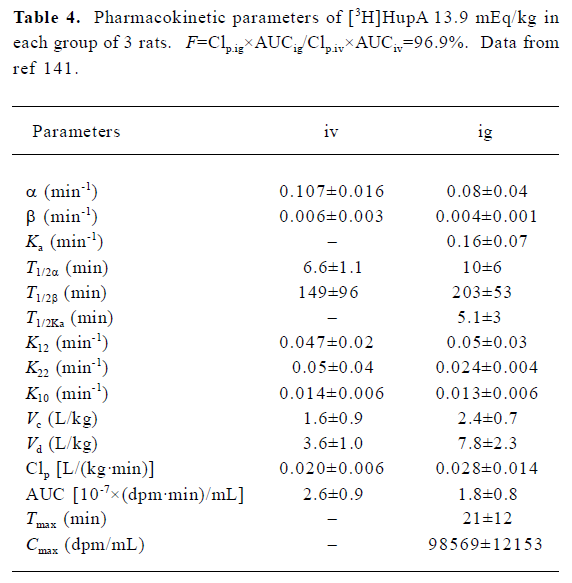
The pharmacokinetics of HupA have been studied in rodents and healthy human volunteers. HupA is absorbed rapidly, distributed widely in the body, and eliminated at a moderate rate (Table 4). The levels of HupA in the blood following iv or po administration of [3H]HupA in rats declined in 2 phases, namely the distribution phase and elimination phase. The oral bioavailability was 96.9% in mice, with the highest radioactivities in the kidney and liver. The majority of the radioactivity was excreted in the urine 24 h after iv administration of [3H]HupA. Only 2.4% was recovered from the feces. Paper chromatograms of rat urine revealed that [3H]HupA was excreted partially as prototype and its metabolite[141]. Autoradiographic studies in mice showed that HupA was present in all regions of the brain, but was particularly concentrated in the frontoparietal cortex, striatal cortex, hippocampus, and nucleus accumbens after iv injection[32].
Full table
The plasma concentration-time curves of HupA in dogs were determined by using the liquid chromatography- mass spectrum-mass spectrum (LC-MS-MS) method after the last intramuscular injection (10 µg/kg per day for 15 d) of a sustained-release formulation, and the mean Cmax was 0.36±0.08 ng/mL, which occurred at 48.0±24.5 h. The mean plasma elimination half-life was 54.8±5.6 h, and the mean area under the plasma concentration versus time curve was 92.6±4.5 ng· h/mL[142]. A pharmacokinetic study using HupA transdermal patches in 6 beagle dogs showed that the HupA patches were able to deliver sustained or controlled drug release in vivo. Following application of the first patch (4 mg per 20 cm2), HupA concentrations in serum increased for approximately 12–24 h, reaching an average maximum concentration of 3.4 ng/mL. Thereafter, blood concentrations were maintained up to 84 h during the period in which the patches were worn[143].
In young healthy volunteers, HupA levels in plasma were determined by reverse phase high performance liquid chromatography (HPLC) by using a spectrophotometric detector. The time course of plasma concentration conformed to a one-compartment open model with first-order absorption following oral administration of 0.99 mg HupA. HupA was rapidly absorbed and widely distributed in vivo[144]. The half-life of HupA was at least 4–17 times longer than that of tacrine or physostigmine[145]. A pharmacokinetic comparison between young and elderly human volunteers treated with 0.225 mg HupA was recently conducted. The areas under the blood level-time curve (AUC0→36) was 75.0 and 97.2 nmol/L·h in young and elderly volunteers, respectively. The mean maximum blood concentrations (Cmax) were 9.1 and 6.8 nmol/L, respectively, and the elimination half-lives (T1/2) of HupA were 10.0 and 13.0 h, respectively. Thus the mean residence times (MRT0–72) were 10.3 and 12.2 h, respectively (Chen Y et al, unpublished data). The elimination of HupA in the elderly volunteers was slower than that in the young volunteers. HupA was released in a slow and prolonged manner after oral administration.
To identify which cytochrome P450 (CYP) isoenzymes are involved in the metabolism of HupA, an in vitro study was performed with rat liver microsomes, and immunoinhibi-tion and chemical inhibition methods. HupA metabolism was analyzed with HPLC and expressed as the HupA disappearance rate. The results showed that 76.2% of HupA metabolism was inhibited by a CYP1A2 antibody and 17.8% by a CYP3A1/2 antibody. The inhibitory effects produced by CYP2C11 and 2E1 antibodies were minor. The CYP1A2 substrate phenacetin produced an inhibitory effect of 70.3%. These data suggest that HupA metabolism in rat liver microsomes is mediated primarily by CYP1A2, with a probable secondary contribution by CYP3A1/2. CYP2C11 and 2E1 are probably not involved in HupA metabolism[146].
To predict possible drug interactions and confirm the safety of HupA as a medication, the effects of HupA on the activity and expression of CYP were examined. Liver microsomes and total mRNA were prepared from rats treated orally with 0, 0.1, 1, or 2 mg/kg HupA for 2 weeks. Phenobarbital, 3-methylcholanthrene (3-MC), ethanol, and dexamethasone were used as positive controls. No change in isoenzyme expression or catalytic activity was found in rats treated with 0.1 mg/kg HupA, but CYP1A2 activity and levels of CYP1A2 protein and mRNA were increased when treated with HupA at doses of 1 and 2 mg/kg, although they were minor when contrasted with 3-MC. HupA produced no effects on CYP2C11, CYP2B1/2, 2E1, or 3A. These results indicate that the activity and expression of liver CYP isoenzymes are not affected in rats treated with pharmacological doses of HupA, but may elicit a slight inductive response in CYP1A2 at a toxicological dose. The CYP1A2 induction produced by HupA is related to transcription enhancement[147].
Toxicology and detoxification
A series of studies have been conducted to evaluate the toxicity of HupA in mice, rats, rabbits, and dogs. Dose-response curves for salivation indicated that HupA was less potent than other ChE inhibitors[49]. The characteristic symptoms of cholinergic hyperactivity were less severe for HupA in rats compared with donepezil and tacrine; fasciculation or other cholinergic signs were not found with oral HupA at a dose of 0.48 mg[19]. The LD50 doses of HupA were 4.6 mg (po), 3.0 mg (sc), 1.8 mg (ip) and 0.63 mg (iv) in mice. Atropine exerted a significant antagonistic effect on the toxicity induced by HupA. Studies to evaluate subacute toxicity have been conducted in rats, rabbits, and dogs, in which no histopathological changes were found in liver, kidney, heart, lung, or brain in rats (1.5 mg/kg, po) or dogs (0.6 mg/kg, im) after administration of HupA for 180 d. No teratogenic effect was detected in mice (0.019–0.38 mg/kg, ip) or rabbits (0.02–0.2 mg/kg, im) after the administration of HupA.
To examine the acute effects of HupA on rat liver, changes in liver coefficient, serum biochemistry, and histopathology were detected after a single dose. The cytotoxicity of HupA was also assessed by determining extracellular and intracellular amounts of lactate dehydrogenase in cultured hepato-cytes. Similar to tacrine, HupA raised the liver coefficient and increased serum levels of aspartate aminotransferase and alanine aminotransferase. Unlike tacrine, however, acute administration of HupA did not induce histopathological changes in the liver. That atropine redressed the effects of HupA on the liver indicates that the acute effects of HupA on rat liver are not related to hepatotoxicity[148].
A study in mice also demonstrated that HupA did not perturb respiration at a dose inhibiting 40% of AChE, and at a lethal dose, did not affect any other enzyme important for respiration[149].
HupA has been tested as a prophylactic drug against poisoning with soman and other nerve gases with reasonable outcomes[150]. It works by protecting cortical AChE from soman inhibition and preventing subsequent seizures.
Several studies have reported that acute administration of HupA protects rodents against organophosphate (OP) intoxication without the typical cholinergic side-effects[150–152]. Similarly, in primates (Chinese rhesus monkeys), HupA by itself has been shown to protect animals against the toxic signs and lethality induced by the injection of 1.3 LD50 of soman[151]. When compared with pyridostigmine (PYR)[153,154], the cumulative dose of soman needed to produce convulsions and epileptic activity was 1.5-fold greater in animals that received HupA compared with the group of primates pretreated with PYR. HupA also selectively inhibited red cell AChE activity, whereas PYR also inhibited plasma BuChE. This result was confirmed in a study with guinea-pigs[155]. PYR cannot protect against seizures and subsequent neuropathology induced by OP agents, because it does not penetrate into the brain. HupA, when combined with atropine methyl nitrate or without any supporting therapy acting on the CNS (atropine sulfate or benzodiazepine), prevented lethality and manifested anticonvulsant and central neuropro-tective properties[155]. The superior protection offered by HupA appears to be related both to the selectivity of HupA for red cell AChE, which preserves the scavenger capacity of plasma BuChE for OP agents, and to its protective effect on cerebral AChE[154]. HupA is more stable than the carbamates used as pretreatment for OP poisoning. These prophylactic effects make HupA a potential protective agent against OP intoxication.
Clinical trials
The efficacy and safety of HupA in AD patients have been evaluated by clinical trials in China. In an early double-blinded study conducted in 100 elderly patients with 17 probable cases of AD, HupA (0.03 mg, im) produced a significant improvement in all rating scores as evaluated by Buschke Selective Reminding performance[156]. A more comprehensive clinical study was conducted in 819 patients who met the AD criteria of National Institute for Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) and Diagnostic and Statistic Manual of Mental Disorders-Third Edition-Revised (DSM-III-R) at 39 mental hospitals in China. After treatment with HupA at a dose of 0.03–0.4 mg/d, patients showed improvement in their memory, cognitive skills, and ability in their daily life. No severe side effects were found[14–16,157–170]. The results from a 12-week, double-blinded, randomized and placebo-controlled nationwide clinical trial with 202 patients with the diagnosis of possible or probable AD confirmed the efficacy of HupA in improving the results of cognitive tasks. In this study, their cognitive functions [measured with Mini-mental State Examination Scale (MMSE) and Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog)], non-cognitive functions (measured with mood and behavior-ADAS-non-Cog) and activity of daily living (ADL) were all improved significantly at week 6, and further improved at week 12 after patients had been treated with HupA at a dose of 0.1–0.2 mg, bid[17]. The results of a clinical trial with HupA carried out over a longer time period were reported by Jiang et al, and showed that HupA at a dose of 0.15 mg twice per day significantly improved the cognition of 33 AD patients at 12, 24, and 48 weeks, with no statistical difference among the 3 time points[164].
Combined therapy with HupA plus other medicine or mental training also showed favorable clinical results. In a placebo-controlled clinical trial with 24 AD patients and 35 VD patients, subjects showed significant favorable differences in MMSE and ADL scores after treated with HupA (0.15 mg, twice per day) plus nicergoline (20 mg, twice per day), HupA plus nicergoline and conjugated estrogen, or HupA alone; combined treatment was better than HupA alone[171]. The superiority of combined therapy over HupA alone was also suggested by the marked improvement in MMSE, clinical dementia rating (CDR) and ADL scores from a clinical trial, in which 30 female AD patients were treated with 2 mg nilestriol once every fortnight, and 0.1 mg HupA twice per day for 24 weeks[172]. In a group of 43 patients with mild to moderate AD, American Association of Mental Deficiency (AAMD) and ADL scores were significantly improved after treatment with HupA (0.1 mg, twice per day) combined with training in daily life activities for 8 weeks[173]. After 22 patients with AD and 38 with VD who were treated for 8 weeks with HupA (0.1 mg, twice per day) complemented with a mental stimulation program consisting of reminiscence, reality orientation and remotivation, Hasegawa dementia scale (HDS), CDR and ADL scores were significantly improved[174]. These results suggest that combined treatment using HupA might be a reasonable means in the clinical therapy.
The therapeutic effects of HupA on VD were evaluated early in 1991 by conducting a randomized, matched and double-blinded study involving 56 patients with multi-infarct dementia (MID)[175]. Using a self-controlled design, marked improvement in memory deficits were observed after 0.15–0.45 mg HupA given orally for 4 weeks[176,177]. Patients with AD (23 cases) or VD (41 cases) were treated with HupA for 8 weeks, and memory deficiency and recognition decline was markedly improved in both AD and VD[178]. Yin et al reported that 39 patients who met the DSM-R criteria for mild to moderate vascular dementia, showed a significant increase in AAMD, MMSE and ADL scores after treated with 0.1 mg HupA twice per day for 8 weeks[179]. A similar result was reported in a trial involving 20 patients[180]. Moreover, HupA has been shown to be more effective than pyritinol in the treatment of MID[181]. Wang et al reported that the CDR and ADL scores of VD patients who met the DSM-IV criteria were markedly improved in MMSE after treatment for 6 months with HupA[20]. HupA also showed efficacy in improving cognitive deficiency in endemic cretins[182], and urinary incontinence in patients with cerebral stroke[183].
A survey involving 50 middle-aged and elderly patients with different degrees of dysmnesia demonstrated the efficacy of HupA in improving verbal recall, retention and retrieval in patients with mild and moderate dysmnesia. Values of total recall, long-term retrieval, long-term storage, consistent long-term retrieval and unreminded recall were markedly increased when patients were treated with 0.1 mg HupA orally twice daily for 2 weeks[184]. In a study with conducted in children who had language delay and other developmental conditions, treatment with 0.05 mg HupA twice daily for more than 3 months improved language delay by a total of 67.56%[185].
Zhang et al reported that HupA could improve memory function and neurotransmission in patients with mild and moderate traumatic brain injury. Thirty patients were treated with traditional therapeutic (0.8 g piracetan and 20 mg nimodipine, twice per day, combined with function convalescence training), and other 30 patients were treated with 0.1 mg HupA bid besides traditional therapeutic. Both groups showed significant improvement in memory and cognition after both 1 and 3 months, and the improvement in memory and cognition in patients treated with HupA was more dramatic than the improvement in patients treated with traditional therapeutics[186].
It is well documented that people with schizophrenia have neurocognitive impairments across multiple domains, including impairments in motor functioning, various aspects of attentional abilities, executive functions and memory functioning. Ma et al have recently studied the effect of HupA on memory disorders in schizophrenic patients. Sixty patients with schizophrenia, who were at the rehabilitation stage, were divided into groups that received HupA or a placebo, and 30 non-schizophrenic people were used as controls. After 12 weeks of treatment with 0.2–0.4 mg HupA, memory functions were significantly improved[187]. Similar results were also reported by Fang et al[188] and Yang[189].
HupA has also shown some efficacy in ameliorating sleeping. It was reported that alternating HupA treatment with clonazepam treatment in the day and night, respectively, markedly extends sleeping duration, modifies the rhythm of sleep, and improves the quality of sleeping in patients with chronic insomnia. With this treatment regime, the dosage of clonazepam can be decreased gradually[190].
Several clinical studies have shown that HupA is effective for the treatment of benign senescent forgetfulness (BSF)[156,175,191–193], in addition to AD and VD. Seventy four percent of patients treated with HupA (0.15 mg, bid) for 4 weeks showed improvement in memory quotient (MQ) and Wechsler memory scale (WMS) scores[15,194–197]. The improving effect of HupA was studied in 34 pairs of junior high school students who had complained of memory inadequacy. HupA at a dose of 0.1 mg, twice per day for 4 weeks, increased the scores for “accumulation”, “recognition”, “reproduction”, “association”, “tactual memory”, and “number of recitation” factors, but not the “understanding” factor[170].
In the early clinical studies using HupA for the treatment of myasthenia gravis (MG), it was found that 99% of 128 patients with MG had their clinical manifestations controlled or improved with HupA treatment. The duration of action of HupA lasted for 7±6 h, and the side effects were minimal compared with neostigmine[198]. Xia et al reported similar improving effects in 63 MG patients treated with im HupA at a dose of 0.2 mg twice per day[199].
Summary
AD is a multi-causal and multi-factorial progressive neurodegenerative disease with complicated pathogenesis, thus it is likely that multiple drugs or drugs with poly-pharmacological activities will be the best therapeutic approaches to address the varied pathological aspects of the disease. Based on the characteristic cholinergic deficits in AD, AChE inhibitors are still the drugs of choice for the symptomatic therapy of AD. As a potent and reversible AChE inhibitor, HupA has attracted attention because, relative to other well-known AChE inhibitors, it has greater potency, higher selectivity with respect to its AChE inhibitory effect, and marked memory-enhancing efficacy in a broad range of animal models of cognitive impairment, and in patients with AD, VD, and other cognitive problems. Interestingly, new data from our lab show that HupA, besides inhibiting the hydrolysis of synaptic ACh, has neuroprotection, APP metabolism modulation and NGF-like neurotrophic activities, indicating that the non-cholinergic effects of HupA could play important roles for the treatment of neurodegenerative disease through interfering with the key factors of the disease. These encouraging preclinical and clinical findings suggest that HupA is a promising candidate for the treatment of neurodegenera-tive diseases such as AD and VD, and is very likely to exert its therapeutic effects via a multi-target mechanism, which therefore provides us with a large amount of exciting research to carry out.
References
- Bartus RT, Dean RL III, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982;217:408-14.
- Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. BMJ 1978;2:1457-9.
- Giacobini E. Cholinesterase inhibitors for Alzheimer’s disease therapy: from tacrine to future applications. Neurochem Int 1998;32:413-9.
- Kumar V. Introduction to cholinesterase inhibitors used in Alzheimer’s disease therapy. In: Giacobini E, Becker R, editors. Alzheimer’s disease: therapeutic strategies. Boston: Birkhäuser; 1994. p 99–102.
- Ellis JM. Cholinesterase inhibitors in the treatment of dementia. J Am Osteopath Assoc 2005;105:145-58.
- Francis PT, Nordberg A, Arnold S. A preclinical view of cholinesterase inhibitors in neuroprotection: do they provide more than symptomatic benefits in Alzheimer’s disease? Trends Pharmacol Sci 2005;26:104-11.
- Farlow M, Gracon SI, Hershey LA, Lewis KW, Sadowsky CH, Dolan-Ureno J. A controlled trial of tacrine in Alzheimer’s disease. JAMA 1992;268:2523-9.
- Knapp MJ, Knopman DS, Solomon PR, Pendlebury WW, Davis CS, Gracon SL. A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer’s disease. JAMA 1994;271:985-91.
- Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology 1998;50:136-45.
- Wang T, Tang XC. Reversal of scopolamine-induced deficits in radial maze performance by (–)-huperzine A: comparison with E2020 and tacrine. Eur J Pharmacol 1998;349:137-42.
- Wang YE, Yue DX, Tang XC. Anticholinesterase activity of huperzine A. Acta Pharmacol Sin 1986;7:110-3. Chinese..
- Zhao Q, Tang XC. Effects of huperzine A on acetylcholinesterase isoforms in vitro: comparison with tacrine, donepezil, rivastigmine and physostigmine. Eur J Pharmacol 2002;455:101-7.
- Liang YQ, Tang XC. Comparative effects of huperzine A, donepezil and rivastigmine on cortical acetylcholine level and acetylcholinesterase activity in rats. Neurosci Lett 2004;361:56-9.
- Xu SS, Gao ZX, Weng Z, Du ZM, Xu WA, Yang JS, et al. Efficacy of tablet huperzine A on memory, cognition, and behavior in Alzheimer’s disease. Acta Pharmacol Sin 1995;16:391-5.
- Xu SS, Xie HB, Du ZW, Tong ZH, Shi QC, Lu KM, et al. Efficacy of tablet huperzine A on memory and cognition in patients with benign senescent forgetfulness. Chin J Clin Pharmacol Ther 1997;2:1-4. Chinese..
- Xu SS, Cai ZY, Qu ZW, Yang RM, Cai YL, Wang GQ. Huperzine A in capsules and tablets for treating patients with Alzheimer’s disease. Acta Pharmacol Sin 1999;20:486-90.
- Zhang ZX, Wang XD, Chen QT, Shu L, Wang JZ, Shan GL. Clinical efficacy and safety of huperzine alpha in treatment of mild to moderate Alzheimer disease, a placebo-controlled, double-blind, randomized trial. Natl Med J China 2002;82:941-4. (Chinese).
- Ellman GL, Courtney KD, Andre V Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88-95.
- Wang H, Tang XC. Anticholinesterase effects of huperzine A, E2020, and tacrine in rats. Acta Pharmacol Sin 1998;19:27-30.
- Wang RP, Zhao ZK, Hu LL. Effects of huperzine A on the cognition and daily life ability in vascular dementia: 36 cases for 6 months follow-up study. Chin J Clin Rehabil 2004;8:3892. Chinese..
- Ogura H, Kosasa T, Kuriya Y, Yamanishi Y. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find Clin Pharmacol 2000;22:609-13.
- Cheng DH, Ren H, Tang XC. Huperzine A, a novel promising acetylcholinesterase inhibitor. Neuoreport 1996;8:97-101.
- Gong ZH, Qin BY. The inhibitory characteristics of fordine on cholinesterases. Bull Acad Mil Med Sci 1986;10:451-6. Chinese..
- Hao XY, Gong ZH, Qin BY. Effects of huperzine A on cholinesterase isoenzymes in plasma of mice and dogs. Acta Pharmacol Sin 1988;9:312-6. Chinese..
- Darvesh S, Arora RC, Martin E, Magee D, Hopkins DA, Armour JA. Cholinesterase inhibitors modify the activity of intrinsic cardiac neurons. Exp Neurol 2004;188:461-70.
- Cheng DH, Tang XC. Comparative studies of huperzine A, E2020, and tacrine on behavior and cholinesterase activities. Pharmacol Biochem Behav 1998;60:377-86.
- Brimijoin S. Molecular forms of acetylcholinesterase in brain, nerve and muscle: nature, localization and dynamics. Prog Neurobiol 1983;21:291-322.
- Massoulie J, Bon S. The molecular forms of cholinesterase and acetylcholinesterase in vertebrate. Annu Rev Neurosci 1982;5:57-106.
- Bon S, Vigny M, Massoulie J. Asymmetric and globular forms of AChE in mammals and birds. Proc Natl Acad Sci USA 1979;76:2540-50.
- Grassi J, Vigny M, Massoulie J. Molecular forms of acetylcholinesterase in bovine caudate nucleus and superior cervical ganglion: solubility properties and hydrophobic character. J Neurochem 1982;387:457-69.
- Tang XC, De Sarno P, Sugaya K, Giacobini E. Effect of huperzine A, a new cholinesterase inhibitor, on the central cholinergic system of the rat. J Neurosci Res 1989;24:276-85.
- Tang XC, Kindel GH, Kozikowski AP, Hanin I. Comparison of the effects of natural and synthetic huperzine A on rat brain cholinergic function in vitro and in vivo. J Ethnopharmacol 1994;44:147-55.
- Laganiere S, Corey J, Tang XC, Wülfert E, Hanin I. Acute and chronic studies with the anticholinesterase huperzine A: Effect on central nervous system cholinergic parameters. Neuropharmacology 1991;30:763-8.
- Zhang HY, Liang YQ, Tang XC, He XC, Bai DL. Stereoselec-tivities of enantiomers of huperzine A in protection against beta amyloid 25–35-induced injury in PC12 and NG108–15 cells and cholinesterase inhibition in mice. Neurosci Lett 2002;317:143-6.
- Raves ML, Harel M, Pang YP, Silman I, Kozilowski AP, Sussman JL. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (–)-huperzine A. Nat Strut Biol 1997;4:57-63.
- Dvir H, Jiang HL, Wong DM, Harel M, Chetrit M, He XC, et al. X-ray structures of Torpedo californica acetylcholinesterase complexed with (+)-huperzine A and (–)-huperzine B: structural evidence for an active site rearrangement. Biochemistry 2002;41:10810-8.
- Ashani Y, Peggins JO III, Doctor BP. Mechanism of inhibition of cholinesterase by huperzine A. Biochem Biophys Res Commun 1992;184:7719-26.
- Pang YP, Kozikowski AP. Prediction of the bonding site of huperzine A in acetylcholinesterase by docking studies. J Computer Aided Mol Des 1994;8:669-81.
- Skolnick AA. Old Chinese herbal medicine used for fever yields possible new Alzheimer disease therapy. JAMA 1997;277:776.
- Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C, et al. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc Natl Acad Sci USA 1993;90:9031-5.
- Saxena A, Qian N, Kovach IM, Kozikowski AP, Pang YP, Vellom DC, et al. Identification of amino acid residues involved in the binding of huperzine A to cholinesterase. Protein Sci 1994;3:1770-8.
- Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, et al. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 1991;253:872-9.
- Lin JH, Hu GY, Tang XC. Facilitatory effect of huperzine A on mouse neuromuscular transmission in vitro. Acta Pharmacol Sin 1996;17:299-301.
- Lin JH, Hu GY, Tang XC. Comparison between huperzine A, tacrine and E2020 on cholinergic transmission at mouse neuromuscular junction in vitro. Acta Pharmacol Sin 1997;18:6-10.
- Zhang GB, Wang MY, Zheng JQ, Tang XC. Facilitation of cholinergic transmission by huperzine A in toad paravertebral ganglia in vitro. Acta Pharmacol Sin 1994;15:158-61.
- Mo N, Dun NJ, Karczmar AG. Facilitation and inhibition of nicotine transmission by eserine in the sympathetic ganglia of the rabbit. Neuropharmacology 1985;24:1093-101.
- Wang MY. Enhancement and depression of cholinergic transmission by 9-amino-1,2,3,4-tetrahydroacridine in rat superior cervical ganglia. Chin Pharmacol Bull 1993;9:298-300. Chinese..
- Fayuk D, Yakel JL. Regulation of nicotinic acetylcholine receptor channel function by acetylcholinesterase inhibitors in rat hippocampal CA1 interneurons. Mol Pharmacol 2004;66:658-66.
- Yan XF, Lu WH, Lou WJ, Tang XC. Effects of huperzine A and B on skeletal muscle and electroencephalogram. Acta Pharmacol Sin 1987;8:117-23. Chinese..
- Guan LC, Chen SS, Cui QG, Lu WH, Tang XC. The effect of huperzine A on behavior and ECoG in animals. Acta Psychol Sin 1991;23:404-11.
- Guan LC, Chen SS, Lu WH, Tang XC. The effect of huperzine A on behavior and ECoG in animals. Acta Pharmacol Sin 1991;12:496-500. Chinese..
- Patil KD, Buerki RA, Patil PN. Potentiation of acetylcholine action by huperzine-A and physostigmine on some vertebrate effectors, including human iris sphincter muscle. J Ocul Pharmacol Ther 2003;19:135-43.
- De Sarno P, Pomponi M, Giacobini E, Tang XC, Williams E. The effect of heptyl-physostigmine, a new cholinesterase inhibitor, on the central cholinergic system of the rat. Neurochem Res 1989;14:971-7.
- Nordberg A, Nilsson L, Adem A, Hardy J, Winblad B. Effect of THA on acetylcholine release and cholinergic receptor in Alzheimer brains. In: Giacobini E, Becker R, editors. Current research in Alzheimer therapy. New York: Taylor and Francis; 1988. p 247–58.
- Hallak M, Giacobini E. Physostigmine, tacrine and metrifonate. The effect of multiple doses on acetylcholine metabolism in rat brain. Neuropharmacology 1989;28:199-206.
- Zhu XD, Giacobini E. Second generation cholinesterase inhibitors: effect of (L)-huperzine A on cortical biogenic amines. J Neurosci Res 1995;41:828-35.
- Bowen DM, Allen SJ, Benton JS, Goodhardt MJ, Haan EA, Palmer AM, et al. Biochemical assessment of serotonergic and cholinergic dysfunction and cerebral atrophy in Alzheimer’s disease. J Neurochem 1983;41:266-72.
- Lu WH, Shou J, Tang XC. Improving effect of huperzine A on discrimination performance in aged rats and adult rats with experimental cognitive impairment. Acta Pharmacol Sin 1988;9:11-5. Chinese..
- Tang XC, Han YF, Chen XP, Zhu XD. Effects of huperzine A on learning and retrieval process of discrimination performance in rats. Acta Pharmacol Sin 1986;7:507-11. Chinese..
- Zhu XD, Tang XC. Facilitatory effects of huperzine A and B on learning and memory of spatial discrimination in mice. Acta Pharmacol Sin 1987;22:812-7. Chinese..
- Zhu XD, Tang XC. Improvement of impaired memory in mice by huperzine A and huperzine B. Acta Pharmacol Sin 1988;9:492-7. Chinese..
- Gao Y, Tang XC, Guan LC, Kuang PZ. Huperzine A reverses scopolamine- and muscimol-induced memory deficits in chick. Acta Pharmocol Sin 2000;21:1169-73.
- Liu J, Zhang HY, Tang XC, Wang B, He XC, Bai DL. Effects of synthetic (–)-huperzine A on cholinesterase activities and mouse water maze performance. Acta Pharmacol Sin 1998;19:413-6.
- Ye JW, Shang YZ, Wang ZM, Tang XC. Huperzine A ameliorates the impaired memory of aged rat in the Morris water maze performance. Acta Pharmacol Sin 2000;21:65-9.
- Xiong ZQ, Tang XC. Effect of huperzine A, a novel acetylcholinesterase inhibitor, on radial maze performance in rats. Pharmacol Biochem Behav 1995;51:415-9.
- Ye JW, Cai JX, Wang LM, Tang XC. Improving effects of huperzine A on spatial working memory in aged monkeys and young adult monkeys with experimental cognitive impairment. J Pharmacol Exp Ther 1999;288:814-9.
- Ou LY, Tang XC, Cai JX. Effect of huperzine A on working memory in reserpine- or yohimbine-treated monkeys. Eur J Pharmacol 2001;433:151-6.
- Vincent GP, Rumennik L, Cumin R, Martin J, Sepinwall J. The effects of huperzine A, an acetylcholinesterase inhibitor, on the enhancement of memory in mice, rats and monkeys. Neurosci Abs 1987;13:844.
- Xiong ZQ, Han YF, Tang XC. Huperzine A ameliorates the spatial working memory impairments induced by AF64A. Neuroreport 1995;6:2221-4.
- Zhang C, Wang SZ, Zuo PP, Cui X, Cai J. Protective effect of tetramethylpyrazine on learning and memory function in D-galactose-lesioned mice. Chin Med Sci J 2004;19:180-4.
- Tang XC, Xiong ZQ, Qian BC, Zhou ZF, Zhang CC. Cognitive improvement by oral huperzine A: a novel acetylcholinesterase inhibitor. In: Giacobini E, Becker R, editors. Alzheimer therapy: therapeutic strategies. Boston: Birkhäuser; 1994. p 113–9.
- Xiong ZQ, Cheng DH, Tang XC. Effects of huperzine A on nucleus basalis magnocellularis lesion-induced spatial working memory deficit. Acta Pharmacol Sin 1998;19:128-32.
- Coyle JT, Price DL, Delong MR. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science 1983;219:1184-90.
- Walsh TJ, Stackman RW. Modulation of memory by benzodiazepine-acetylcholine interactions. In: Butcher LL, Decker MW, Levin ED, editors. Neurotransmitter interactions and cognitive function. Boston: Birkhäuser; 1992. p 312–28.
- Wenk GL, Pierce DJ, Struble RG, Price DL, Cork LC. Aged-related changes in multiple neurotransmitter systems in the monkey brain. Neurobiol Aging 1989;10:11-9.
- Wang R, Zhang HY, Tang XC. Huperzine A attenuates cognitive dysfunction and neuronal degeneration caused by beta-amyloid protein-(1–40) in rat. Eur J Pharmacol 2001;21:149-56.
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 1993;361:31-9.
- Sucher NJ, Awobuluyi M, Choi YB, Lipton SA. NMDA receptors: from genes to channels. Trends Pharmacol Sci 1996;17:348-55.
- Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res 2000;60:65-72.
- Ye L, Qiao JT. Suppressive action produced by beta-amyloid peptide fragment 31–35 on long-term potentiation in rat hippocampus is N-methyl-D-aspartate receptor-independent: it’s offset by (–)-huperzine A. Neurosci Lett 1999;275:187-90.
- Wang LM, Han YF, Tang XC. Huperzine A improves cognitive deficits caused by chronic cerebral hypoperfusion in rats. Eur J Pharmocol 2000;398:65-72.
- Zhou J, Zhang HY, Tang XC. Huperzine A attenuates cognitive deficits and hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci Lett 2001;313:137-40.
- Wang LS, Zhou J, Shao XM, Tang XC. Huperzine A attenuates cognitive deficits and brain injury in neonatal rats after hypoxiaischemia. Brain Res 2002;949:162-70.
- Butterfield DA, Howard B, Yatin S, Koppal T, Drake J, Hensley K, et al. Elevated oxidative stress in models of normal brain aging and Alzheimer’s disease. Life Sci 1999;65:1883-92.
- Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 1997;23:134-47.
- Selkoe DJ, Abraham CR, Podlisny MB, Duffy LK. Isolation of low-molecular-weight proteins form amyloid plaque fibers in Alzheimer’s disease. J Neurochem 1986;46:1820-34.
- Gilgun-Sherki Y, Melamed E, Offen D. Antioxidant treatment in Alzheimer’s disease: current state. J Mol Neurosci 2003;21:1-12.
- Xiao XQ, Yang JW, Tang XC. Huperzine A protects rat pheochromocytoma cells against hydrogen peroxide-induced injury. Neurosci Lett 1999;275:73-6.
- Xiao XQ, Wang R, Han YF, Tang XC. Protective effects of huperzine A on β-amyloid25–35 induced oxidative injury in rat pheochromocytoma cells. Neurosci Lett 2000;286:155-8.
- Xiao XQ, Wang R, Tang XC. Huperzine A and tacrine attenuate β-amyloid peptide induced oxidative injury. J Neurosci Res 2000;61:564-9.
- Xiao XQ, Zhang HY, Tang XC. Huperzine A attenuates amyloid β-peptide fragment 25–35-induced apoptosis in rat cortical neurons via inhibiting reactive oxygen species formation and caspase-3 activation. J Neurosci Res 2002;67:30-6.
- Zhou J, Fu Y, Tang XC. Huperzine A and donepezil protect rat pheochromocytoma cells against oxygen-glucose deprivation. Neurosci Lett 2001;306:53-6.
- Shang YZ, Ye JW, Tang XC. Improving effects of huperzine A on abnormal lipid peroxidation and superoxide dismutase in aged rats. Acta Pharmacol Sin 1999;20:824-8.
- Lü PY, Yin Y, Wang WB, Liang CP, Li WB. Effects of huperzine A on [Ca2+]i level and expression of CaM, CaMPK II mRNA in hippocampal neurons of mice with vascular dementia. Clin J New Drugs Clin Rem 2004;23:73-6.
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature 2000;407:802-9.
- Wang R, Xiao XQ, Tang XC. Huperzine A attenuates hydrogen peroxide-induced apoptosis by regulation expression of apoptosis-related genes in rat PC12 cells. Neuroreport 2001;12:2629-34.
- Zhou J, Fu Y, Tang XC. Huperzine A protects rat pheochromocytoma cells against oxygen-glucose deprivation. Neuroreport 2001;12:2073-7.
- Zhou J, Tang XC. Huperzine A attenuates apoptosis and mitochondria-dependent caspase-3 in rat cortical neurons. FEBS Lett 2002;526:21-5.
- Zhang HY, Tang XC. Huperzine A attenuates the neurotoxic effect of staurosporine in primary rat cortical neurons. Neurosci Lett 2003;340:91-4.
- Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev 1996;10:1054-72.
- Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie 2000;84:153-66.
- Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 2003;304:463-70.
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 1998;17:37-49.
- Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s Box opens. Nat Rev Mol Cell Biol 2001;2:67-71.
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997;278:114-7.
- Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW. Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol Dis 1998;5:81-8.
- Malin SA, Guo WX, Jafari G, Goate AM, Nerbonne JM. Presenilins upregulate functional K+ channel currents in mammalian cells. Neurobiol Dis 1998;4:398-409.
- Li Y, Hu GY. Huperzine A, a nootropic agent, inhibits fast transient potassium current in rat dissociated hippocampal neurons. Neurosci Lett 2002;324:25-8.
- Li Y, Hu GY. Huperzine A inhibits the sustained potassium current in rat dissociated hippocampal neurons. Neurosci Lett 2002;329:153-6.
- Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci 1994;747:162-71.
- DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci 1990;13:286-9.
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999;22:391-7.
- Hossmann KA. Glutamate-mediated injury in focal cerebral ischemia: the excitotoxin hypothesis revised. Brain Pathol 1994;4:23-36.
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int 2004;45:583-95.
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 1994;330:613-22.
- Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci 1988;11:465-9.
- Choi DW. Calcium: still center-stage in hypoxic–ischemic neuronal death. Trends Neurosci 1995;18:58-60.
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature 1999;399 Suppl 6738:A7-14.
- Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 1984;226:850-2.
- Turski L, Huth A, Sheardown M, McDonald F, Neuhaus R, Schneider HH, et al. ZK200775: a phosphonate quinoxaline-dione AMPA antagonist for neuroprotection in stroke and trauma. Proc Natl Acad Sci USA 1998;95:10960-5.
- Ved HS, Koening ML, Dave JR, Doctor BP. Huperzine A, a potential therapeutic agent for dementia, reduces neuronal cell death caused by glutamate. NeuroReport 1997;8:963-8.
- Gordon RK, Nigam SV, Weitz JA, Dave JR, Doctor BP, Ved HS. The NMDA receptor ion channel: a site for binding of huperzine A. J Appl Toxicol 2001;21 Suppl 1:S47-51.
- Wang XD, Zhang JM, Yang HH, Hu GY. Modulation of NMDA receptor by huperzine A in rat cerebral cortex. Acta Pharmacol Sin 1999;20:31-5.
- Zhang JM, Hu GY. Huperzine A, a nootropic alkaloid, inhibits N-methyl-D-aspartate-induced current in rat dissociated hippocampal neurons. Neuroscience 2001;105:663-9.
- Zhang YH, Zhao XY, Chen XQ, Wang Y, Yang HH, Hu GY. Spermidine antagonizes the inhibitory effect of huperzine A on [3H]dizocilpine (MK-801) binding in synaptic membrane of rat cerebral cortex. Neurosci Lett 2002;319:107-10.
- Zhang YH, Chen XQ, Yang HH, Jin GY, Bai DL, Hu GY. Similar potency of the enantiomers of huperzine A in inhibition of [(3)H]dizocilpine (MK-801) binding in rat cerebral cortex. Neurosci Lett 2000;295:116-8.
- Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, et al. Cleavage of amyloid beta-peptide during constitutive processing of its precursor. Science 1990;248:1122-4.
- Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid-beta peptide. Cell 1993;75:1039-42.
- Gasparini L, Racchi M, Binetti G, Trabucchi M, Solerte SB, Alkon D, et al. Peripheral markers in testing pathophysiological hypotheses and diagnosing Alzheimer’s disease. FASEB J 1998;12:17-34.
- Racchi M, Govoni S. Rationalizing a pharmacological intervention on the amyloid precursor protein metabolism. Trends Pharmacol Sci 1999;20:418-23.
- Nitsch RM, Slack BE, Wurtman RJ, Growdon J. Release of Alzheimer amyloid precursor derivative stimulated by activation of muscarinic acetylcholine receptors. Science 1992;258:304-7.
- Giacobini E, Mori F, Lai CC. The effect of cholinesterase inhibitors on the secretion of APPs from rat brain cortex. Ann NY Acad Sci 1996;777:393-8.
- Mori F, Lai CC, Fusi F, Giacobini E. Cholinesterase inhibitors increase secretion of APPs in rat brain cortex. NeuroReport 1995;6:633-6.
- Zhang HY, Yan H, Tang XC. Huperzine A enhances the level of secretory amyloid precursor protein and protein kinase C-α in intracerebroventricular β-amyloid-(1–40) infused rats and human embryonic kidney 293 Swedish mutant cells. Neurosci Lett 2004;360:21-4.
- Hefti F, Hartikka J, Knusel B. Function of neurotrophic factors in the adult and aging brain and their possible use in the treatment of neurodegenerative disease. Neurobiol Aging 1989;10:515-33.
- Mufson EJ, Bothwell M, Kordower JH. Loss of nerve growth factor receptor-containing neurons in Alzheimer’s disease: a quantitative analysis across subregions of the basal forebrain. Exp Neurol 1989;105:221-32.
- Shigeta K, Ootaki K, Tatemoto H, Nakanishi T, Inada A, Muto N. Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by a Coptidis Rhizoma extract and protoberine alkaloids. Biosci Biotech Biochem 2002;66:2491-4.
- Brimijoin S, Koenigsberger C. Cholinesterases in neural development: new findings and toxicologic implications. Environ Health Perspect 1999;107 Suppl 1:59-64.
- Tang LL, Wang R, Tang XC. Effects of huperzine A on secretion of nerve growth factor in cultured rat cortical astrocytes and neurite outgrowth in rat PC12 cells. Acta Pharmacol Sin 2005;26:673-8.
- Tang LL, Wang R, Tang XC. Huperzine A protects SHSY5Y neuroblastoma cells against oxidative stress damage via nerve growth factor production. Eur J Pharmacol 2005;519:9-15.
- Wang YE, Feng J, Lu WH, Tang XC. Pharmacokinetics of huperzine A in rats and mice. Acta Pharmacol Sin 1988;9:193-6. Chinese..
- Wang Y, Chu D, Gu J, Fawcett JP, Wu Y, Liu W. Liquid chromatographic-tandem mass spectrometric method for the quantitation of huperzine A in dog plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2004;803:375-8.
- Ye J, Zeng S, Zhang W, Chen G. Ion-pair reverse-phase high performance liquid chromatography method for determination of huperzine-A in beagle dog serum. J Chromatogr B Analyt Technol Biomed Life Sci 2005;817:187-91.
- Qian BC, Wang M, Zhou ZF, Chen K, Zhou RR, Chen GS. Pharmacokinetics of tablet huperzine A in six volunteers. Acta Pharmacol Sin 1995;16:396-8.
- Hartvig P, Wiklund L, Aquilonius SM, Lindström B. Clinical pharmacokinetics of centrally acting cholinesterase inhibitors. In: Becker R, Giacobini E, editors. Cholinergic basis for Alzheimer therapy. Boston: Birkhäuser; 1991. p 68–73.
- Ma XC, Wang HX, Xin J, Zhang T, Tu ZH. Indentification of cytochrome P450 1A2 as enzyme involved in the microsomal metabolism of huperzine A. Eur J Pharmacol 2003;461:89-92.
- Ma XC, Wang HX, Xin J, Zhang T, Tu ZH. Effects of huperzine A on liver cytochrome P-450 in rats. Acta Pharmacol Sin 2003;24:831-5.
- Ma XC, Xin J, Wang HX, Zhang T, Tu ZH. Acute effects of huperzine A and tacrine on rat liver. Acta Pharmacol Sin 2003;24:247-50.
- Boudinot E, Taysse L, Daulon S, Chatonnet A, Champagnat J, Foutz AS. Effects of acetylcholinesterase and butyrylcholine-sterase inhibition on breathing in mice adapted or not to reduced acetylcholinesterase. Pharmacol Biochem Behav 2005;80:53-61.
- Grunwald J, Raveh L, Doctor P, Ashani Y. Huperzine A as a pretreatment candidate drug against nerve agent toxicity. Life Sci 1994;54:991-7.
- Ashani Y, Grundwald J, Alkalai D, Cohen G, Raveh L. Studies with huperzine A, a new candidate in the research of prophylaxis against nerve agent. In: King JM, editor. Proceedings of the Medical Defence Bioscience Review 1996. p 105–10.
- Tonduli L, Testylier G, Masqueliez C, Lallement G, Monmaur P. Effects of huperzine used as pretreatment against soman-induced seizures. Neurotoxicology 2001;24:276-85.
- Lallement G, Baille V, Baubichon D, Carpentier P, Collombet JM, Filliat P, et al. Review of the value of huperzine as pretreatment of organophosphate poisoning. Neurotoxicology 2002;23:1-5.
- Lallement G, Demoncheaux JP, Foquin A, Baubichon D, Galonnier M, Clarencon D, et al. Subchronic compared efficacy against soman toxicity. Drug Chem Toxicol 2002;25:309-20.
- Lallement G, Veyret V, Masqueliez M, Aubriot S, Burckhart MF, Baubichon D. Efficacy of huperzine in preventing soman-induced seizures, neuropathological changes and lethality. Fund Clin Pharmacol 1997;11:387-94.
- Zhang CL. Therapeutic effects of huperzine A on the aged with memory impairment. New Drugs Clin Remedies 1986;5:260-2. Chinese..
- Liu FG, Fang YS, Gao ZX, Zhou JD, Su ML. Double-blind control treatment with huperzine A and placebo in 28 patients with Alzheimer disease. Clin J Pharmacoepidemiol 1995;4:196-8. Chinese..
- Yang JS, Jiang ZH. Clinical report of huperzine A for the treatment of Alzheimer’s disease. He Bei Jing Shen Wei Sheng 1996;9:84-5. Chinese..
- Zhao CY, Li SF, Chen XL, Han MK. Efficacy of tablet huperzine A on treating 21 patients with Alzheimer’s disease. Si Chuan Jing Shen Wei Sheng 1996;9:204. Chinese..
- Wang LJ, Ji XX, Weng QS, Yang JS. Clinical observation of huperzine A on 36 patients with Alzheimer’s disease. Nan Tong Yi Xue Yuan Xue Bao 1998;18:486-8. Chinese..
- Liu JN, Huang ZY, Zhou YD, Ju YL. The clinical observation of Alzheimer’s disease treated by huperzine A. Chin J Clin Phar 1998;7:270-2. Chinese..
- Wang LJ, Ji XX, Weng QS, Yang JS. Clinical observation of huperzine A on 36 patients with Alzheimer’s disease. Shanghai Yi Yao 1999;20:16-8. Chinese..
- Chen YM, Ma YX, Chen MJ, Fang YS, Chai XS, Weng Z. Observation of aniracetam and huperzine on memory function of Alzheimer’s disease. Modern Rehabil 2000;4:1622-3. Chinese..
- Jiang YB, Huang SS, Huang LA. Improvement of huperzine A on the deficiency of cognitive and behavior in Alzheimer’s disease. Clin Med China 2002;18:802-3. Chinese..
- Song ZY, Lu H. Clinical efficacy of huperzine A on Alzheimer disease. Chin J Clin Rehabil 2003;7:112. Chinese..
- Yang CY, Lv ZP, Zheng CG. Efficacy and reliability of huperzine A in mild and moderate Alzheimer’s disease. Chin J Clin Rehabil 2003;7:4258-9. Chinese..
- Kuang MZ, Xiao WM, Wang SF, Li RX. Clinical evaluation of huperzine A in improving intelligent disorder in patients with Alzheimer’s disease. Chin J Clin Rehabil 2004;8:1216-7. Chinese..
- Wu SF, Xie SZ, Lu WJ, Ma YX. Comparison of huperzine A in capsule and tablet on random, double-blind treating mild and moderate Alzheimer’s disease. Shanghai Yi Yao 1999;20:36-7. Chinese..
- Chen MJ, Gao ZX, Deng HY, Liu FG, Ma YX, Yu HZ, et al. Huperzine A capsules vs tablets in treatment of Alzheimer disease: multi-center studies. Chin J New Drugs Clin Remedies 2000;19:10-2. Chinese..
- Sun QQ, Xu SS, Pan JL, Guo HM, Cao WQ. Huperzine-A capsules enhance memory and learning performance in 34 pairs of matched adolescent students. Acta Pharmacol Sin 1999;20:601-3.
- Zhou BR, Xu ZQ, Kuang YF, Deng YH, Liu ZF. Effectiveness of polydrug therapy for senile dementia. Chin J Clin Rehabil 2004;8:1214-5. Chinese..
- Wang RQ, Lei QY, Gu JQ, Wang XY, Liu YL, Fang SY, et al. Nilestriol combined with huperzine in improving cognition of female patients with Alzheimer’s disease. Chin J Clin Rehabil 2003;7:1538-9. Chinese..
- Miao XR. Huperzine A assisted with the training of daily life promotes rehabilitation of Alzheimer’s disease. Chin J Clin Rehabil 2002;6:2551. Chinese..
- Wang RQ, Meng HY, Liu W. Therapeutic effects of huperzine A complementing with 3R mental stimulation program in senile dementia patients. Chin J Clin Rehabil 2002;6:2560-1. Chinese..
- Zhang RW, Tang XC, Han YY, Sang GW, Zhang YD, Ma YX, et al. Drug evaluation of huperzine A in the treatment of senile memory disorders. Acta Pharmacol Sin 1991;12:250-2. Chinese..
- Sun CY, Chen XY. A self-control study of huperzine A on memory deficits in patients with multiple infarctions. Henan Yi Yao Xin Xi 1998;6:31-2. Chinese..
- Sun CY, Cai ZH, Chen XY. A self-control study of huperzine A on memory deficits in patients with multiple infarctions. Zhong Yuan Jing Shen Yi Xue Xue Kan 1998;4:151-3. Chinese..
- Ye Q, Wu RZ, Su BZ, Li HL. A study of the efficacy of huperzine A in the treatment of memory deficiency and recognition decline of cerebral organic disease. Sichuan Jing Shen Wei Sheng 2001;14:75-6. Chinese..
- Yin FM, Du YY, Wang LE. The effects of huperzine A on vascular dementia. Modern Rehabil 2001;5:74-5. Chinese..
- Chen YP, Mei YW, Cheng SQ. Treatment on 20 cases of multi-infarct dementia with huperzine A. Yi Yao Dao Bao 2002;21:275-6. Chinese..
- Chang JJ. Huperzine A vs pyritinol in treating dementia from multiple infarctions. New Drugs Clin Remedies 1997;16:333-4. Chinese..
- Qu CY, Wang HM, Yu W, Xue ZW. A pilot trial of huperzine A on treating the cognitive deficiency in endemic cretinism. Shanxi Yi Yao Za Zhi 1995;24:47-8. Chinese..
- Yang XY, Zhang HY. Therapeutic effects of huperzine A on urinary incontinence after cerebral stroke. J Pract Nerv Dis 2004;7:77. Chinese..
- Chang SY, Chen SM, Cao QL, Liu P, Wang FG, Wang ZX. A clinical study of the effect of huperzine A to improve the ability of verbal recall, retention and repetition in middle-aged and elderly patients with dysmnesia. Herald Med 2002;21:263-5. Chinese..
- Liao JX, Chen L, Huang TS. Pilot trial of huperzine A to treat child language delay. J Pediatr Pharm 2002;8:26-7. Chinese..
- Zhang JH, Fan JZ, Deng AW. A clinical study of huperzine A on mild and moderate traumatic brain injury in memory and cognitive impairment. Chin J Rehabil Med 2002;17:162-4. Chinese..
- Ma JD, Zheng H, Wang YJ. Effect of huperzine A on the memory disorders of schizophrenic patients during rehabilitation period. Health Psychol J 2003;11:340-1. Chinese..
- Fang CX, Guo CR, Wu B, Jing YT. Effects of huperzine A on memory patients with schizophrenia. Shandong Jing Shen Yi Xue 2002;15:39-40. Chinese..
- Yang JZ. The effects of huperzine A on the cognitive deficiency of rehabilitating schizophrenia. Chin J Clin Rehabil 2003;7:1440. Chinese..
- Gao X, Yu QP, Cao QH. A clinical trial of day and night alternated administration of huperzine A and clonazepam to treat chronic insomnia. Chin J Nerv Ment Dis 2003;29:58-9. Chinese..
- Zhang CL, Wang GZ. Effects of huperzine A tablet on memory. New Drugs Clin Remedies 1990;9:339-41. Chinese..
- Zhang XQ, Ding MC, Meng C, Yang PJ. Oral administration of huperzine A improved memory disorder: a clinical study. Zhong Guo Xin Yao Za Zhi 1996;5:33-4. Chinese..
- Du ZM, Li SL, Yang CF, Wang YY, Zhang HZ, Xu SS. Double-blind and placebo controlled study of huperzine A on treatment of benign senescent forgetfulness. Chin J Geriatr 1996;15:180. Chinese..
- Zhu XY, Gu YD, Wang HH. Comparison of the efficacy between tablet and capsule of huperzine to treat age associated memory impairment by double blind trial method. Zhong Guo Lin Chuang Yao Xue Za Zhi 1999;8:227-9. Chinese..
- Li SL, Xu SS. Huperzine A capsules vs tablets in treating senile amnesia: a double-blind study. Chin J New Drugs Clin Remedies 2000;19:33-5. Chinese..
- Chai XS, Sheng JH. Huperzine A capsule vs huperzine A in treatment of age-associated memory impairment with a double-blind study. Shandong Arch Psychiatr 2001;14:88-90. Chinese..
- Sheng JH, Gao ZX, Chai XS, Zhou F, Cui SS. Huperzine A capsule in treatment of age-associated memory impairment with a double-blind study. Sichuan Jing Shen Wei Sheng 2003;16:1-3. Chinese..
- Cheng YS, Lu CZ, Ying ZL, Ni WY, Zhang CL, Sang GW. One hundred and twenty-eight cases of myasthenia gravis treated with huperzine A. New Drugs Clin Remedies 1986;5:197-9.
- Xia Q, Liu QC. Clinical study of huperzine A on the treatment of myasthenia gravis. Proc J Med Pharm 2002; 19: 31. Chinese.