
Intracellular dopamine oxidation mediates rotenone-induced apoptosis in PC12 cells1
Introduction
Parkinson disease (PD) is a chronic neurodegenerative disorder characterized by the loss of dopamine (DA) neurons in the substantia nigra, decreased striatal DA levels, and consequent extrapyramidal motor dysfunction. Although the genes responsible for a few rare familial cases of PD have been discovered[1-3], the causes of more prevalent idiopathic PD are still unknown. Substances that are toxic to dopaminergic cells have been proposed as contributors to this neurological disease[4,5]. MPTP (1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is one of the best described neuro-toxins. Evidence has shown that the neurotoxicity of MPTP depends on its active metabolite, 1-methyl-4-phenyl-pyri-dinium (MPP+), which inhibits mitochondrial complex I and depletes cellular ATP levels, resulting in cell death[6]. Thus, one hypothesis has focused on mitochondrial dysfunction[7].
Rotenone, a naturally occurring, lipophilic compound from the roots of certain plants (Derris species), is a specific inhibitor of mitochondrial complex I and is used as the main component of many insecticides. Based on the mitochondrial dysfunction hypothesis of PD, a number of studies have evaluated the effects of rotenone on dopaminergic neurons both in vitro and in vivo[8-15]. Treatment of mesencephalic cultures and striatal synaptosomes with rotenone caused neurotoxicity which was measured by a decreased uptake of neurotransmitters[8,9]. Studies in vivo have shown that rotenone was capable of causing degeneration of dopaminergic neurons and induction of parkinsonian symptoms in animals[10-15]. An important morphological finding in rotenone-treated rats was that the nigrostriatal DA neurons had accumulated fibrillar cytoplasmic inclusions containing ubiquitin and alpha-synuclein, similar to Lewy bodies in idiopathic PD[13,16]. This finding is interesting, because recent discoveries of the two causative gene products of familial PD, Parkin[1,17,18] and alpha-synuclein[3] indicate that failure of the ubiquitin-proteasome system might be common in both familial PD and idiopathic PD[19]. Another reason is that rotenone is widely used as insecticide, and therefore is a real threat as an environmental substance to cause PD. Thus, elucidation of its mode of action is of high importance in understanding and potentially treating this disorder. The present study was designed to assess the neurotoxicity of rotenone on DA-producing PC12 cells and explore the possible mechanism.
Materials and methods
Reagents Dulbecco’s modified Eagle’s medium (DMEM) was purchased from GibcoBRL (Gaithersburg, MD, USA). Horse serum and fetal calf serum were obtained from Hyclone (Logan, UT, USA). Rotenone, reserpine, deprenyl, N-acetyl-L-cysteine (NAC), propidium iodide (PI), dihydroxybenzylamine (DHBA) and 2´,7´-dichlorofluo-rescein-diacetate (DCFH-DA) were purchased from Sigma (St Louis, MO, USA). Caspase-3 substrate AC-L-aspartic-L-glutamic-L-alyl-L-aspartic acid-7-amino-4-methylcou-marin (Ac-DEVD-AMC) was obtained from Calbiochem (La Jolla, CA, USA).
Cell culture The rat pheochromocytoma cell line PC12 cells (American Type Culture Collection, Rockville, MD, USA) were propagated in DMEM, supplemented with heat-inactivated horse serum (10%, v/v) and fetal calf serum (5%, v/v), 100 g/L streptomycin and 100 kU/L penicillin. The cultures were maintained in an incubator at 37 oC in a high humidity atmosphere of 5% CO2. The medium was changed every 2 d and cells were passaged once a week. Twenty-four hours before addition of various reagents, the cells were seeded on 60-mm-dishes (Falcon), covered with collagen (Sigma), at a density of 1×105/cm2 in normal medium. Rotenone and reserpine were first dissolved in dimethyl sulfoxide (Me2SO) and then diluted in medium to a final concentration of Me2SO less than 0.1%. Controls for each drug condition consisted of sister cultures treated with the vehicle used to dissolve that drug. Stock solutions of deprenyl and NAC were prepared in Hanks’ balanced salt solution containing 2 mmol/L HEPES.
Assessment of cell viability Cell viability was assessed by detecting the leakage of lactate dehydrogenase (LDH) into the medium. Cells were treated with rotenone or other reagents, then an aliquot of medium was taken and centrifuged at 250×g for 5 min. Supernatant (10 mL) was added into phosphate buffer 0.1 mol/L (pH 7.0) containing sodium pyruvate 2.3 mmol/L and NADH 5 mmol/L to a total volume of 200 mL. The decrease in absorbance over time at 340 nm was monitored at 25 ºC. Then the activity of LDH in medium was calculated. Cell total LDH was obtained after exposure of culture to 0.2% TritonX-100 at 37 ºC for 30 min. LDH leakage was expressed as percentage of LDH in medium to total LDH.
Analysis of apoptosis by flow cytometryApoptosis rate was measured by flow cytometry as reported previously[19]. Briefly, PC12 cells were washed with PBS (pH 7.4), fixed in cold 70% (v/v) ethanol, and incubated under -20 oC for at least 2 h. The fixed cells were harvested by centrifugation at 250×g for 5 min. The cell pellets were resuspended in 1 mL PBS at room temperature for 10 min. After another centrifugation, the cell pellets were resuspended in 500 mL PBS containing 0.2 g/L RNase A and incubated at 37 ºC for 30 min. After incubation, the cells were stained with 20 g/L PI at 4 ºC for 30 min. The fluorescence of cells was measured with FACSCalibur flow cytometer (BD Immunocyto-metry Systems, San Jose, CA, USA). The relative content of DNA indicated the distribution of a population of cells throughout the cell cycle. Apoptotic cells caused the appearance of a sub-diploid peak in the cell-cycle profile. The percentage of apoptotic cells was determined by using BD CellQuest software.
Assay for caspase-3 activityCaspase-3-like activity was measured as described in a previous study with modification[20]. In brief, the PC12 cells were collected and washed with PBS (pH 7.4). After centrifugation at 250×g for 5 min, cell pellets were lysed with NP-40 (0.5%)/HEPES (10 mmol/L) (pH 7.4), containing EDTA 2 mmol/L, PMSF 0.5 mmol/L and leupeptin 5 mg/L. The lysates were centrifuged at 7500×g for 10 min. The protein concentration in the supernatant was determined with Lowry method. Then 50 µg of protein was incubated with caspase-3 substrate Ac-DEVD-AMC 50 µmol/L. The increase of fluorescence was measured every 1 min in a 30 min period using a PolarStar plate reader (BMG labtechnologies, Australia) with an excitation wavelength of 380 nm and an emission wavelength of 450 nm. The enzyme activity was expressed as fluorescent units per min per mg protein.
Measurement of reactive oxygen species productionThe level of intracellular hydrogen peroxide and other peroxides in PC12 cells were quantified by loading cells with 2´-7´-dichlorodihydrofluorescein diacetate (DCFH-DA) as described previously[19]. In brief, the PC12 cells were washed with PBS (pH 7.4), resuspended in Krebs-Ringer buffer (HEPES 20 mmol/L, dextrose 10 mmol/L, NaCl 127 mmol/L, CaCl2 1 mmol/L, KCl 5.5 mmol/L, MgSO4 2 mmol/L, pH 7.4), and then loaded with DCFH-DA 5 mmol/L for 60 min. The cells were again washed with Krebs-Ringer buffer to remove the extracellular dye and lysed in Tris-HCl 10 mmol/L (pH 7.4) containing 0.5% Triton X-100. The fluorescence in lysates was detected using a PolarStar plate reader (BMG labtechnologies, Australia) with an excitation wavelength of 485 nm and an emission wavelength of 520 nm. The fluorescence intensity was normalized based on the protein concentration of individual extract. The ROS production was expressed as percentage compared with cells without treatment.
Determination of lactic acid accumulation Lactic acid accumulation was measured as described previously[21]. In brief, PC12 cells were homogenized with sodium phosphate 50 mmol/L (pH 6.5) and boiled for 15 min. Lactic acid content was measured spectrophotometrically at 340 nm in a buffer containing LDH and hydrazine (0.4 mol/L)/glycine (0.5 mol/L) (pH 9.0). Lactic acid accumulation was normalized based on the protein concentration of individual extract, and expressed as percentage compared with cells without treatment. Protein concentration was detected by the Lowry method.
Measurement of dopamine in PC12 cellsThe DA levels in PC12 cells were determined by a modification of the methods described previously[22-24]. In brief, cells were lysed in 1% metaphosphoric acid containing EDTA 1 mmol/L. Each sample added with 20 mL of DHBA was regarded as an internal standard. After centrifugation (17 500×g for 10 min at 4 ºC), the supernatant was filtered, and a 20 mL aliquot was immediately injected into the HPLC system for DHBA and DA determination. The assay was done with a BAS PM80 pump, a Waters Nova-Pak C18 column, and an electrochemical detector (BAS LC-4C). The mobile phase was citric acid 0.1 mol/L, K2HPO4 0.1 mol/L, EDTA 0.1 mmol/L, 5% methanol, and sodium octylsulfate 70 mg/L (pH 3.0); the flow rate was 1.0 mL/min. The potential of the electrode was set at +0.7 V. DA level in each sample was quantified by comparing DA and DHBA peak areas with those of standard solutions containing DA and DHBA, and then normalized based on the protein concentration of each individual sample. The DA level was expressed as percentage compared with control cells without reserpine treatment.
Statistics Data for cell viability, caspase-3 activity, ROS production and lactic acid accumulation were presented as mean±SD of four independent experiments. Data for dopamine levels in cells are means±SD of three experiments performed in triplicate. Statistical analysis was performed by applying the Student’s t-test and one-way ANOVA.
Results
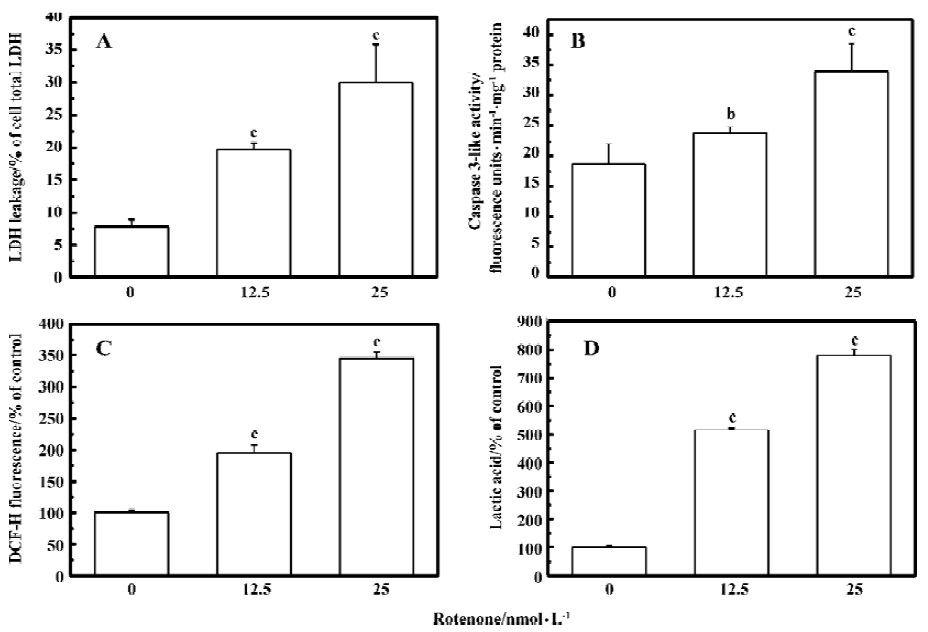
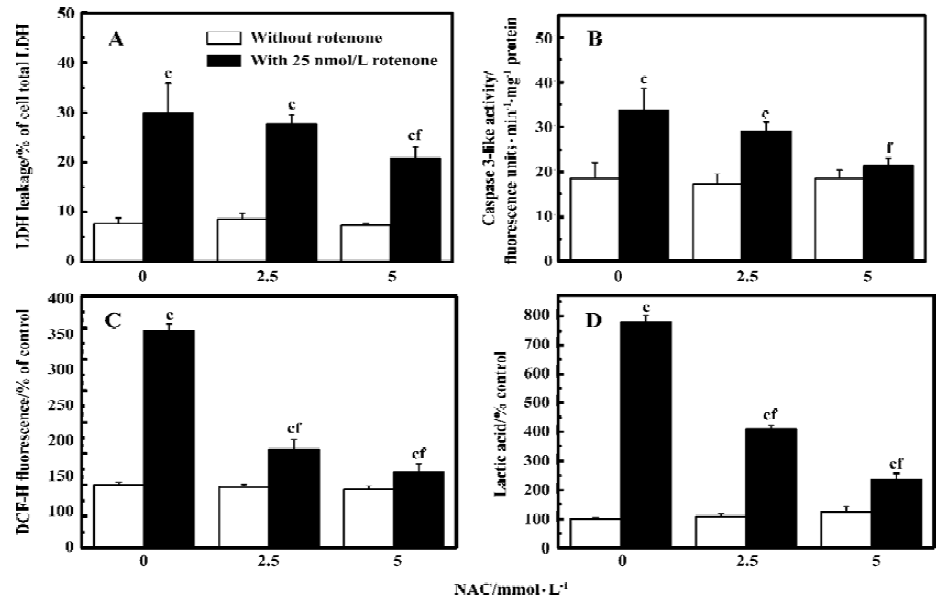
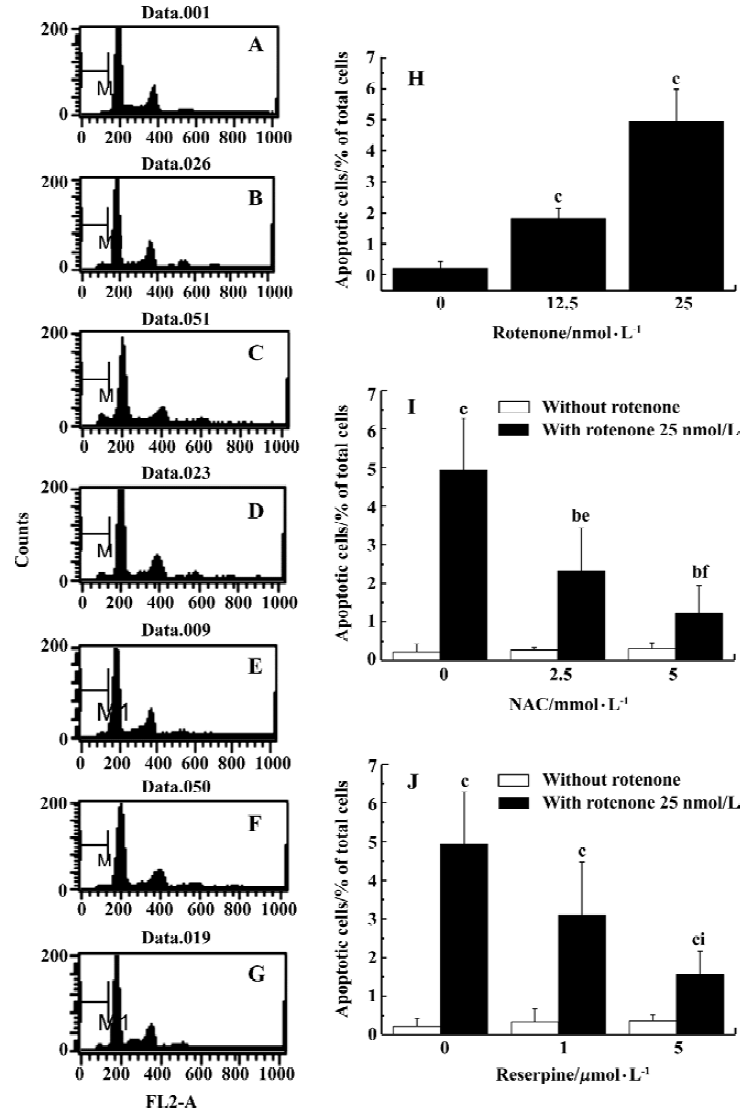
Oxidative stress is involved in rotenone-induced neurotoxicity When PC12 cells were treated with 12.5 and 25 nmol/L rotenone for 48 h, LDH leakage increased from 7.8%±1.1% to 19.7%±1.1% and 30.1%±3.6% (P<0.01 vs control), respectively (Figure 1A). The rotenone-induced cell injury was associated with caspase-3 activation (Figure 1B), supporting the notion that an apoptotic cell death mechanism was involved in rotenone-induced neurotoxicity. Rotenone treatment induced a concentration-dependent increase of ROS levels within PC12 cells (Figure 1C). Rotenone treatment also induced an increase in lactic acid accumulation in PC12 cells (Figure 1D), suggesting that both oxidative stress and mitochondrial dysfunction were responsible for cell injury. The effects of rotenone were attenuated by 2.5 and 5 mmol/L NAC co-treatment (Figure 2A-2C), further supporting the notion that oxidative stress was involved in rotenone neurotoxicity. In addition, 2.5 and 5 mmol/L NAC also attenuated rotenone-induced increase in lactic acid accumulation (Figure 2D).
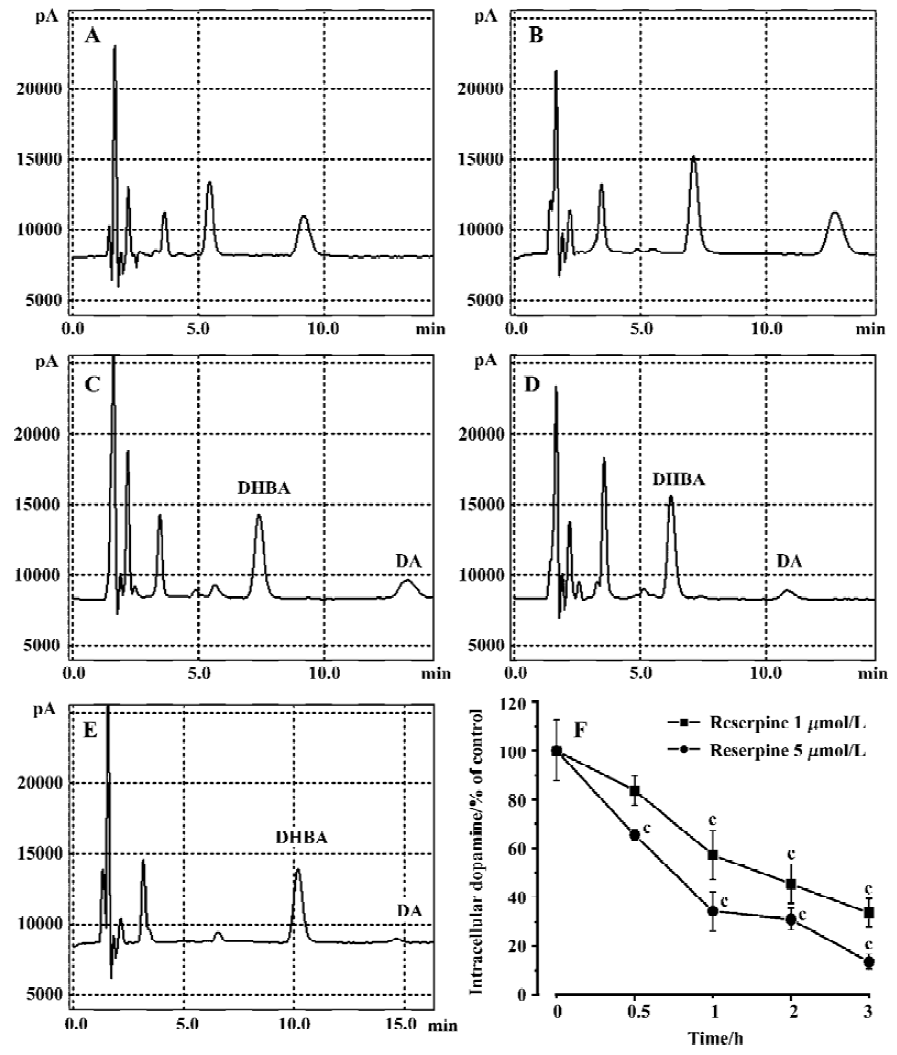
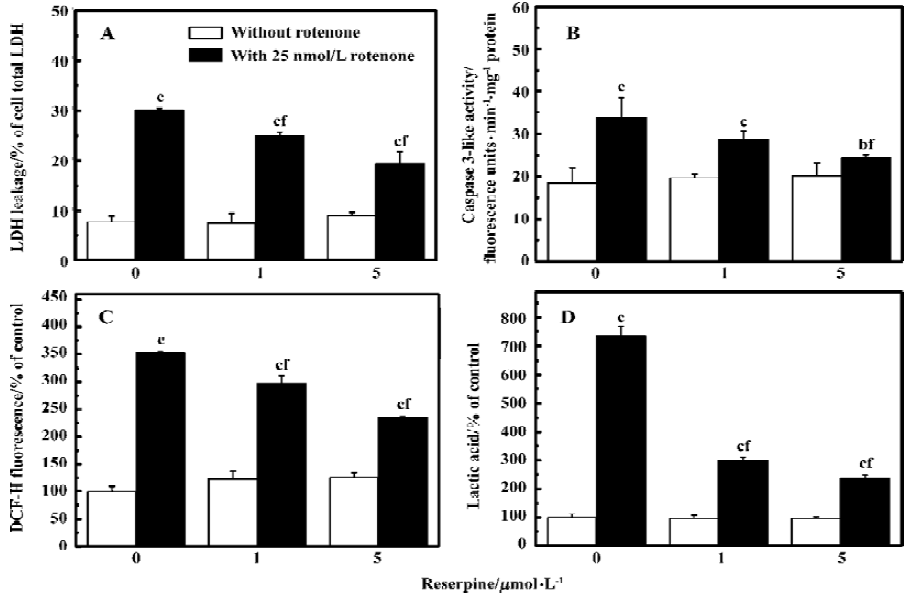
Rotenone-induced ROS accumulation is mediated by DA oxidationTo test whether manipulation of DA levels would attenuate rotenone-induced cell death, PC12 cells were first depleted of intracellular DA using reserpine and then treated with rotenone. Reserpine (1 and 5 µmol/L) treatment for 3 h induced 66% and 87% depletions of DA respectively as compared with the control (Figure 3). The rotenone-induced elevation of LDH leakage and caspase-3 activation were attenuated in reserpine-pretreated PC12 cells (Figure 4A–4B), suggesting that DA played an important role in rotenone-induced cell injury. The rotenone-induced intracellular accumulation of ROS was also attenuated by this manipulation (Figure 4C). The results suggest that DA oxidation is involved in the production of ROS. In addition, lactic acid accumulation assay indicated that the rotenone-induced energy crisis was partially alleviated if PC12 cells were pretreated with reserpine (Figure 4D).
Analyzed quantitatively by flow cytometry, the apoptotic rates increased significantly in PC12 cells treated with rotenone (12.5 and 25 nmol/L) for 48 h (Figure 5A). The co-treatment with NAC 2.5 and 5 mmol/L or pre-treatment with reserpine 1 and 5 mmol/L for 3 h also decreased the rotenone-induced apoptotic rate (Figure 5B and 5C).
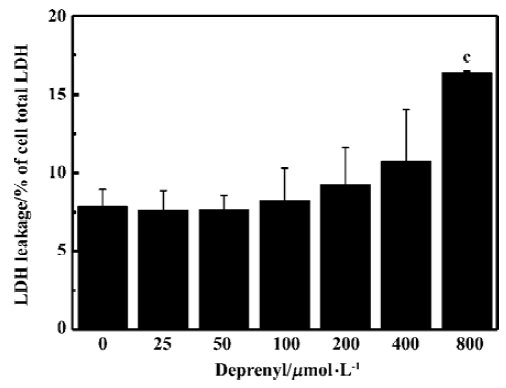
DA auto-oxidation is responsible for rotenone-induced neurotoxicityTo test whether DA deamination or auto-oxidation was more responsible for rotenone-induced neurotoxicity in PC12 cells, deprenyl, a monoamine oxidase-B (MAO-B) inhibitor, was used. The treatment with deprenyl at concentration between 25 to 200 µmol/L for 48 h had no significant effects on the LDH leakage in PC12 cells (Figure 6). When PC12 cells were first treated with 100 µmol/L deprenyl for 30 min and then co-treated with deprenyl 100 µmol/L plus rotenone 25 nmol/L for 48 h, no significant difference was found in the LDH leakage, caspase-3 activation or ROS production if compared with PC12 cells treated with rotenone 25 nmol/L alone (Table 1). The results suggest that the oxidative deamination of DA catalyzed by MAO-B was not a major source of ROS in the present studies.

Full table
Vesicular monoamine transporter is crucial for cell survival when there is mitochondrial dysfunction plus enchanced DA auto-oxidationTo examine whether vesicular monoamine transporter (VMAT) was involved in rotenone-induced neurotoxicity in PC12 cells, 1 or 5 µmol/L reserpine was added 30 min before the end of 48 h-rotenone treatment. No change was found in rotenone-induced neurotoxicity with the brief reserpine treatment. However, when PC12 cells were pretreated with deprenyl 100 µmol/L for 30 min and then with deprenyl 100 µmol/L plus rotenone 25 nmol/L for 48 h, the 30-min incubation with reserpine 1 or 5 µmol/L before the end of 48-h treatment with deprenyl plus rotenone induced significant increases in LDH leakage, caspase-3 activation, and ROS (Table 1).
Discussion
In the present study, the mechanisms of rotenone-induced neurotoxicity in DA-producing PC12 cells was invesigated. We found that rotenone induced an apoptotic type of cell death. This result is similar to the results reported by Bal-Price and Brown[25]. They found that a 24-h incubation of PC12 cells with nitric oxide donors or rotenone, in the presence of glucose, induced apoptosis of PC12 cells as determined by nuclear morphology and caspase-3 activation. We found that rotenone induced apoptosis in PC12 cells accompanied with an elevated ROS production. Given that mitochondria is a major cellular source of ROS[26] and that rotenone is a kind of complex I inhibitor, it could be hypothesized that rotenone-induced ROS might be originated from mitochondria. Indeed, high concentrations of rotenone have been reported to induce superoxide production in vitro and in vivo[27-30]. In isolated nerve terminals, Sipos et al[31] found that inactivation of complex I to a small extent (16%) resulted in a significant increase in ROS formation. However, other studies have suggested converse results[32-34]. Lotharius and O’Malley[35] found that low doses (5-50 nmol/L) of rotenone killed dopaminergic neurons with a similar time course and morphology to MPP+. However, no increase was found in intracellular superoxide levels after the dopaminergic neurons were treated with rotenone for 0.5, 1, 3, and 6 h. Because the classic complex I inhibitor rotenone did not induce ROS, they concluded that MPP+-induced superoxide did not arise from blockade of electron transport. In the present study, however, we found a significant increase of ROS production from PC12 cells after rotenone treatment for 48 h. While DCFH-DA was used as a probe to measure the redox state of a cell, the resluts were validated carefully. We found that the rotenone-induced ROS accumulation and cell death were attenuated by an antioxidant, NAC. This result supports the notion that an oxidative stress mechanism is involved in rotenone-induced neurotoxicity in PC12 cells.
If toxic agents were involved in the pathogenesis of PD, this should likely reflects a multifactorial etiology in which the effects of environmental insults are compounded by predisposing genetic traits, age-related changes and interactions with endogenous elements (i.e. factors inherent to the nigrostriatal tissue). The presence of dopamine within nigro-striatal neurons may itself constitute a risk factor that enhances their vulnerability to toxic events, such as increased production of oxidizing species. To test whether manipulation of DA levels would attenuate rotenone-induced cell death, cells were depleted of intracellular DA using reserpine prior to rotenone treatment. In the present studies, we demonstrated that reserpine induced a concentration- and time-dependent DA depletion in PC12 cells. When PC12 cells were treated with 1 and 5 mmol/L reserpine for 3 h, 66% and 87% decreases in DA levels were found respec-tively, as compared with the control (Figure 3). Our results were similar to those reported by Brautigam et al[36] who reported more that 50% depletion of intracellular DA in PC12 cells after reserpine incubation. Our finding that DA depletion significantly attenuated rotenone-induced ROS production and cell death suggests that DA plays an important role in rotenone-induced ROS production and cell death. Since DA in the cytoplasm can be readily autoxidized[37] or deaminated[38] by MAO-B to produce ROS[39], deprenyl, a MAO-B inhibitor, was used to test whether MAO-B inhibition could protect PC12 cells from rotenone-induced ROS accumulation and cell death. Although it has been shown that MAO-B inhibition by pretreatment with deprenyl significantly reduces cellular DOPAC formation in PC12 cells[40], it had no effect on the rotenone-induced ROS accumulation and cell death in our studies, suggesting that the oxidative deamination of DA catalyzed by MAO-B was not the main source of ROS in rotenone-treated PC12 cells and that DA auto-oxidation is responsible for rotenone-induced oxidative stress.
The presence of dopamine within nigrostriatal neurons can itself constitute a risk factor that enhances their vulnerability to toxic agents. For example, rotenone-induced dendrite loss was severe in the substantia nigra, whereas nonca-techolamine neurons, such as those in the perifornical nucleus, were more resistant[41]. Other factors might also play roles in the vulnerability to toxic agents since some dopamine neurons in hypothalamic A11 and the ventral tegmental area were spared when treated with rotenone. By in situ hybridization, a weaker expression of VMAT was found in the rat substantia nigra than in the ventral tegmental area[42]. If VMAT effectively protects the cell against oxidative stress induced by dopamine, the weak expression of VMAT in substantia nigra might account for its particular vulnerability to toxic agents.
No change was found in the rotenone-induced neurotoxi-city. However, when PC12 cells were pretreated with deprenyl and then with deprenyl plus rotenone for 48 h, significant increases in LDH leakage, caspase-3 activation, and ROS production were found. The results suggest that VMAT activity might be important for cell survival if there is mitochondrial dysfunction. In other words, disruption in dopamine disposition and/or metabolism could underlie the progressive degeneration of dopaminergic neurons in PD.
As a specific inhibitor of mitochondrial complex I, the metabolic consequence of mitochondrial electron flow blockage induced by rotenone would be the incomplete oxidation of glucose and consequent lactic acid accumulation. Indeed, we showed that rotenone treatment also induced an increase in lactic-acid accumulation. The results support the notion that mitochondrial dysfunction is responsible for rotenone-induced cell death in PC12 cells. Mitochondria in PC12 cells can become more vulnerable to hydrogen peroxide-induced oxidative stress when complex I is inhibited.
Taken together, our results demonstrated that both oxidative stress and mitochondrial dysfunction were responsible for rotenone-induced apoptotic cell death in PC12 cells, and that rotenone-induced ROS accumulation was mainly DA auto-oxidation mediated.
Acknowledgment
The authors wish to thank Dr Ren-gang WANG and Ms Li-zhen WANG for their technical suggestions.
Footnote
Work supported by a grant from the Chinese Academy of Science and Grant G (1998) 051108 from the Ministry of Sciences and Technology of China.
References
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605-8.
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998;395:451-2.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045-7.
- Jenner P. Parkinson’s disease, pesticides and mitochondrial dysfunction. Trends Neurosci 2001;24:245-7.
- Sherer TB, Betarbet R, Greenamyre JT. Environment, mitochondria, and Parkinson’s disease. Neuroscientist 2002;8:192-7.
- Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem 1993;61:1191-206.
- Schapira AH. Mitochondrial function and neurotoxicity. Curr Opin Neurol 1994;7:531-6.
- Marey-Semper I, Gelman M, Levi-Strauss M. A selective toxicity toward cultured mesencephalic dopaminergic neurons is induced by the synergistic effects of energetic metabolism impairment and NMDA receptor activation. J Neurosci 1995;15:5912-8.
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, et al. Deficiencies in complex 1 subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 1989;163:1450-5.
- Heikkila RE, Nicklas WJ, Vyas I, Duvoisin RC. Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci Lett 1985;62:389-94.
- Ferrante RJ, Schulz JB, Kowall NW, Beal MF. Systemic administration of rotenone produces selective damage in the striatum and globus pallidus, but not in the substantia nigra. Brain Res 1997;753:157-62.
- Thiffault C, Langston JW, Di Monte DA. Increased striatal dopamine turnover following acute administration of rotenone to mice. Brain Res 2000;885:283-8.
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000;3:1301-6.
- Alam M, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res 2002;136:317-24.
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol 2003;179:9-16.
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa A, Minoshima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000;25:302-5.
- Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, et al. Ubiquitination of a new form of α-synuclein by Parkin from human brain: implications for Parkinson’s disease. Science 2001;293:263-9.
- McNaught KSP, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci 2001;2:589-94.
- Wang RG, Zhu XZ. Subtoxic concentration of manganese synergistically potentiates 1-14 methyl-4-phenylpyridinium-induced neurotoxicity in PC12 cells. Brain Res 2003;961:131-8.
- Zhou LJ, Zhu XZ. Reactive oxygen species-induced apoptosis in PC12 cells and protective effect of Bilobalide. J Pharmacol Exp Ther 2000;293:982-5.
- Hirata Y, Adachi K, Kiuchi K. Activation of JNK pathway and induction of apoptosis by manganese in PC12 cells. J Neurochem 1998;71:1607-15.
- Mefford IN, Giberg M, Barchas JD. Simultaneous determination of catecholamines and unconjugated 3,4-dihydroxyphenylacetic acid (DOPAC) by ion-pairing reverse phase high performance liquid chromatography with electrochemical detection. Anal Biochem 1980;104:469-72.
- Zhu XZ, Luo LG. Effect of nitroprusside (nitric oxide) on endogenous dopamine release from rat striatal slices. J Neurochem 1992;59:932-5.
- Wu WR, Zhu XZ, Guan HJ, Wang RG, Ji XQ. Neuroprotective rather than neurorescue or neurorestorative effect of selegiline against MPTP-induced dopaminergic toxicity. Acta Pharmacol Sin 1999;20:146-50.
- Bal-Price A, Brown GC. Nitric-oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J Neurochem 2000;75:1455-64.
- Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, et al. Oxidants in mitochondria: from physiology to disease. Biochim Biophys Acta 1995;1271:67-74.
- Takeshige K, Minakami S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem J 1979;180:129-35.
- Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 1980;191:421-7.
- Hasegawa E, Takeshige K, Oishi T, Murai Y, Minakami S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun 1990;170:1049-55.
- Packer MA, Miesel R, Murphy MP. Exposure to the parkinsonian neurotoxin 1-methyl-4-phenylpyridinium (MPP+) and nitric oxide simultaneously causes cyclosporin A-sensitive mitochondrial calcium efflux and depolarisation. Biochem Pharmacol 1996;51:267-73.
- Sipos I, Tretter L, Adam-Vizi V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem 2003;84:112-8.
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem 1996;67:2282-91.
- Rottenberg H, Wu S. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim Biophys Acta 1998;1404:393-404.
- Chinopoulos C, Tretter L, Adam-Vizi V. Depolarization of in situ mitochondria due to hydrogen peroxide-induced oxidative stress in nerve terminals: inhibition of alpha-ketoglutarate dehydrogenase. J Neurochem 1999;73:220-8.
- Lotharius J, O’Malley KL. The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem 2000;275:38581-8.
- Brautigam M, Laschinski G, Kittner B, Herken H. Effect of apomorphine, alpha-methylparatyrosine, haloperidol and reserpine on DOPA production in clonal cell lines (PC-12 and N1E-115). Biochem Pharmacol 1985;34:941-7.
- Fornstedt B. Role of catechol autooxidation in the degeneration of dopamine neurons. Acta Neurol Scand Suppl 1990;129:12-4.
- Maker HS, Weiss C, Silides DJ, Cohen G. Coupling of dopamine oxidation (monoamine oxidase activity) to glutathione oxidation via the generation of hydrogen peroxide in rat brain homogenates. J Neurochem 1981;36:589-93.
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol 1978;14:633-43.
- Kitazawa M, Wagner JR, Kirby ML, Anantharam V, Kanthasamy AG. Oxidative stress and mitochondrial-mediated apoptosis in dopaminergic cells exposed to methylcyclo-pentadienyl manganese tricarbonyl. J Pharmacol Exp Ther 2002;302:26-35.
- Bywood PT, Johnson SM. Mitochondrial complex inhibitors preferentially damage substantia nigra dopamine neurons in rat brain slices. Exp Neurol 2003;179:47-59.
- Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, et al. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 1992;70:539-51.