
Novel cyclophilin D inhibitors derived from quinoxaline exhibit highly inhibitory activity against rat mitochondrial swelling and Ca2+ uptake/release
Introduction
Apoptosis is essential for normal development and aging in multicellular organisms, and abnormal regulation of apoptosis can result in multiple human diseases. Mitochondria release apoptogenic proteins such as cytochrome C and apoptosis-inducing factor (AIF) into the cytosol, which are involved in the signaling pathway of caspases and induce cell apoptosis[1–3]. One major pathway of the release of the apoptogenic factors to the cytosol is via the rupture of the outer mitochondrial membrane due to mitochondrial permeability transition (MPT) pore opening[3]. It is suggested that MPT pores play a potent role in cell aging[4,5], and opening of the MPT pores may cause changes in mitochondrial shape and function, such as the massive swelling of mitochondria, rupture of the outer membrane and release of inter-membrane components that induce apoptosis. It has been reported that the agents that inhibit MPT may have therapeutic potential for the treatment of human diseases such as ischemia-reperfusion injury in peripheral organs, trauma and neurodegenerative diseases[5–7].
Recent studies have shown that the MPT pore is composed of three major proteins: the voltage-dependent anion channel (VDAC) in the outer membrane that forms a large H2O-filled pore with a diameter of 2.5–3.0 nm, the adenine nucleotide translocator (ANT) that mediates the ADP-ATP exchange in the inner membrane, and cyclophilin D (CypD)[8]. CypD belongs to the family of highly homologous peptidyl prolyl cis-trans isomerases (PPIases) that are thought to be important for protein folding, and can bind to the immuno-suppressor cyclosporin A (CSA)[9,10]. It is known that CypD is a mitochondrial-targeted PPIase, even though its specific physiological role is largely obscure[11,12]. CypD has been confirmed to play a decisive role in MPT pore regulation, and PPIase activity of CypD might be a necessary step in MPT pore opening[8,13]. A model was recently proposed concerning the mechanism of permeability transition-related cytochrome c release, whereby the Ca2+ requirement for the induction of the MPT pore opening might be due to the Ca2+-dependent interaction between CypD and ANT[14–16]. It has been reported that CypD inhibitor CSA and its analogues may block MPT pore opening[17,18], which thereby makes discovering the CypD inhibitor an appealing project. However, to our knowledge, investigating the small molecular CypD-specific inhibitor for allowing brain penetration is still a challenge.
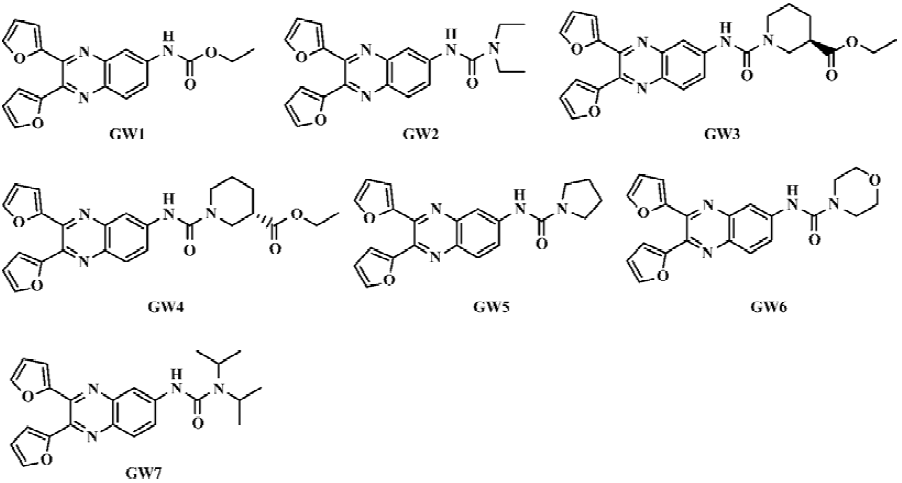
In this paper, we report 7 novel quinoxaline derivatives (Scheme 1 and Figure 1) that inhibit the PPIase activity of CypD. By using surface plasmon resonance (SPR) and fluorescence titration techniques, the kinetics of the CypD-inhibitor interaction was investigated. The compounds’ inhibition effects against rat liver Ca2+-depedent mitochondrial swelling and Ca2+ uptake/release were also determined. The binding selectivity of CypD over CypA for the tested compounds was analyzed, and explained based on the molecular docking technique. We hope that this research will provide a useful approach for the discovery of cyclophilin D inhibitors, and thus help to develop promising compounds using CypD as a drug target for the inhibition of MPT pore opening.
Materials and methods
All solvents and reagents were purchased commercially and were used without further purification. 1H nuclear magnetic resonance (NMR) spectra (400 MHz) were recorded on a Varian (Palo Alto, California, USA) Mercury-400 spectro-meter. Plasmid extraction was performed using the GenElute Plasmid Miniprep Kit (Sigma-Aldrich, St Louis, Missouri, USA).
The compound 6-amino-2,3-di(furan-2-yl)quinoxaline was synthesized according to the patented method[19]. (R)-ethyl nipecotate and (S)-ethyl nipecotate were prepared according to a previously published method[20].
General preparation procedure of compounds GW1–7 The chemical structures of the seven tested compounds are shown in Figure 1, and the general synthetic procedure is shown in Scheme 1. Briefly, the compounds were prepared from 2,4-dinitroaniline in five steps.
2,3-di(furan-2-yl)-6-ethoxycarbonylamino quinoxaline (GW1) To a solution of 6-amino-2,3-di(furan-2-yl)quinoxaline (83.1 mg, 0.30 mmol) and triethylamine (100 µL, 0.72 mmol) in dichloromethane (10 mL) we added triphosgene (30 mg, 0.10 mmol) while stirring. The mixture was stirred at room temperature for 1 h, then ethanol (100 µL, 1.7 mmol) was added. After another 1 h of stirring, the solvent was evaporated in a vacuum to give the crude product, which was further purified by flash column chromatography on a silica gel using petro-ether/ethyl acetate (3:1) as the eluent. The obtained compound GW1 (45.2 mg, 43% yield) was a yellowish amorphous solid. 1HNMR (CDCl3, 400 MHz). d: 8.10 (d, 1H, J=2.4 Hz), 8.05 (d, 1H, J=9.1Hz), 7.87 (dd, 1H, J=2.2 Hz, 9.1 Hz), 7.60 (m, 2H), 7.17 (s, 1H), 6.62 (m, 2H), 5.54 (m, 2H), 4.28 (q, 2H, J=7.0 Hz), 1.32 (t, 3H, J=7.0 Hz); IR (KBr): 3419, 3246, 2982, 2928, 1732, 1622, 1572, 1539, 1495, 1261, 1226 per cm; High-resolution mass spectra(electron impact) calculated value [HRMS(EI) Calcd] for C19H15N3O4 349.1063; Found 349.1068.
2,3-di(furan-2-yl)-6-N-(N',N'-diethylcarbamoyl)amino quinoxaline (GW2) GW2 was synthesized by using a method similar to that described for the preparation of GW1, except that diethylamine was used instead of ethanol. GW2 is a brown amorphous solid (63.0 mg, 56% yield). 1H NMR (CDCl3, 400 MHz). δ: 8.03 (d, 2H, J=1.8 Hz), 7.96 (d, 1H, J=1.0 Hz), 7.59 (dd, 2H, J=0.7 Hz, 1.0 Hz), 6.78 (s, 1H), 6.61 (ddd, 2H, J=0.7 Hz, 3.5 Hz, 12.2 Hz), 6.54 (m, 2H), 3.42 (q, 4H, J=7.2 Hz), 1.25 (t, 6H, J=7.2 Hz); IR (KBr): 3440, 3114, 2972, 2929, 1649, 1524, 1491, 1429, 1265 per cm; HRMS (EI) Calcd for C21H20N4O3 376.1535; Found 376.1528.
2,3-di(furan-2-yl)-6-((R)-3-ethoxycarbonyl-piperidino)carbonylamino quinoxaline (GW3) GW3 was prepared by using a method similar to that described for the preparation of GW1, except that (R)-ethyl nipecotate was used instead of ethanol. GW3 is a yellow amorphous solid (66.4 mg, 48% yield). 1H NMR (CDCl3, 400 MHz). δ: 8.13 (s, 1H), 8.02 (m, 1H), 7.95 (m, 2H), 7.59 (m, 2H), 6.59 (m, 2H), 6.53 (m, 2H), 4.22 (m, 2H), 3.99 (m, 2H), 3.48 (dd, 1H, J=3.3, 14.2 Hz), 3.10 (m, 1H), 2.70 (m, 1H), 2.20 (m, 1H), 1.45–1.95 (m, 3H), 1.29 (t, 3H, J=6.1 Hz); IR (KBr): 3404, 2937, 2860, 1728, 1649, 1570, 1527, 1473, 1431, 1256 cm-1; HRMS(EI) Calcd for C25H24N4O5 460.1747; Found 460.1725. [α]D20 = -52º (c=0.83, CH3OH).
2,3-di(furan-2-yl)-6-((S)-3-ethoxycarbonyl-piperidino)carbonylamino quinoxaline (GW4) GW4 was prepared by using a method similar to that described for GW1 except that (S)-ethyl nipecotate was used instead of ethanol. GW4 was a yellow amorphous solid (68.7 mg, 50% yield). 1H NMR (CDCl3, 400 MHz). δ: 8.13(s, 1H), 8.02 (m, 1H), 7.95 (m, 2H), 7.59 (m, 2H), 6.59 (m, 2H), 6.53 (m, 2H), 4.22 (m, 2H), 3.99 (m, 2H), 3.48 (dd, 1H, J=3.3, 14.2 Hz), 3.10 (m, 1H), 2.70 (m, 1H), 2.20 (m, 1H), 1.45–1.95 (m, 3H), 1.29 (t, 3H, J=6.1 Hz); IR (KBr): 3404, 2937, 2860, 1728, 1649, 1570, 1529, 1475, 1431, 1254 cm-1; HRMS(EI) Calcd for C25H24N4O5 460.1747; Found 460.1738. [α]D20 = +55º (c=1.78, CH3OH).
2,3-di(furan-2-yl) -6-(pyrrolidin-1-yl)carbonylamino quinoxaline (GW5) GW5 was prepared by using a method similar to that described for the preparation of GW1 except that pyrrolidine was used instead of ethanol. GW5 is a brown amorphous solid (106.9 mg, 95% yield). 1H NMR (CDCl3, 400 MHz). δ: 8.18 (dd, 1H, J=2.3, 9.2 Hz), 8.02 (m, 2H), 7.60 (m, 2H), 6.68 (s, 1H), 6.65 (d, 1H, J=3.4 Hz), 6.60 (dd, 1H, J=0.7, 3.4 Hz), 6.55 (m, 2H), 3.53 (t, 4H, J=6.6 Hz), 2.00 (t, 4H, J=6.6 Hz); IR (KBr): 3404, 2970, 2877, 1672, 1568, 1525, 1502, 1429, 1382, 1340, 1203 per cm; HRMS(EI) Calcd for C21H18N4O3 374.1379; Found 374.1360.
2,3-di(furan-2-yl)-6-morpholinocarbonylamino quinoxa-line (GW6) GW6 was prepared by using a method similar to that described for the preparation of GW1 except that morpholine was used instead of ethanol. GW6 was a yellow amorphous solid (101.4 mg, 87% yield). 1H NMR (CDCl3, 400 MHz). δ: 7.96–8.06 (m, 3H), 7.60 (m, 2H), 6.99 (s, 1H), 6.66 (dd, 1H, J=0.8, 3.5 Hz), 6.63 (dd, 1H, J=0.8, 3.5 Hz), 6.56 (m, 2H), 3.77 (t, 2H, J=4.9 Hz), 3.56 (t, 2H, J=4.9 Hz), 1.80 (b, 4H); IR (KBr): 3423, 2920, 2852, 1653, 1529, 1475, 1429, 1333, 1254 per cm; HRMS(EI) Calcd for C21H18N4O4 390.1328; Found 390.1309.
2,3-di(furan-2-yl) -6-N-(N',N'-diisopropylcarbamoyl)amino quinoxaline (GW7) GW7 was prepared by using a method similar to that described for the preparation of GW1 except that diisopropylamine was used instead of ethanol. GW7 is a brown amorphous solid (43.9 mg, 36% yield). 1H NMR (CDCl3, 400 MHz). δ: 7.99–8.06 (m, 2H), 7.90 (d, 1H, J=2.3 Hz), 7.57 (m, 2H), 6.76 (s, 1H), 6.59 (m, 2H), 6.51 (m, 2H), 3.99 (m, 2H), 1.32 (d, 12H, J=6.9 Hz); IR (KBr): 3427, 2968, 2928, 1647, 1566, 1520, 1495, 1433, 1375, 1205 per cm; HRMS(EI) Calcd for C23H24N4O3 404.1848; Found 404.1857.
Preparation of His-tagged human CypA protein All cloning techniques including polymerase chain reaction (PCR), restriction, ligation, E coli transformation, and plasmid DNA preparation were carried out according to standard methods[21]. The His-tagged CypA protein was expressed and purified from the plasmid pQE30-CypA according to the published procedure[22].
Preparation of rat CypD protein The plasmid pcDNA3.1(+)/Zeo-CypD was kindly provided by Dr James D LECHLEITER (University of Texas Health Science Center, U3SA). By using the forward primer 5'-ATAGAATTCATGCT-AGCTCTGCGCTGCG-3' (containing an EcoRI site) and the reverse primer 5'-ATATCTCGAGGCTCAACTGGCCACA-GTC-3' (containing an XhoI site), the PCR product was sub-cloned into the vector pGEX-4T-1 between the EcoRI and XhoI sites to obtain the expression plasmid pGEX-4T-1-CypD. Sequencing was carried out to confirm the insertion.
E coli strains were prepared in Luria-Bertani medium containing 100 mg/mL ampicillin. BL21 (DE3) bacteria transformed with pGEX-4T-1-CypD were grown until the OD600 reached 0.8, and isopropylthio-β-D-galactoside (IPTG) was added to a final concentration of 0.2 mmol/L to induce GST-CypD expression at 25 C overnight.
Bacteria were harvested and lysed by sonication in a sonication buffer [1×phosphate-buffered saline (PBS), 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), pH 7.3, 1 mmol/L ethylenediamine tetraacetic acid (EDTA), 1% Triton X-100]. The bacterial lysate was centrifuged and the supernatant was collected. GST-CypD protein was purified by using a glutathione Sepharose 4B column (Amersham Biosciences, Uppsala, Sweden), and the purified CypD protein was obtained by the on-column cleavage of GST-CypD using thrombin according to the instructions given by the manufacturers of the glutathione Sepharose 4B column (Amersham Biosciences). The purity of the obtained CypD protein was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis as a single band.
Surface plasmon resonance technology-based Biacore 3000 analyses The interactions between compounds GW1–7 and CypD (A) were performed using the dual flow cell Biacore 3000 instrument (Biacore AB, Uppsala, Sweden). All the experiments were carried out using HBS-EP (10 mmol/L N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid [HEPES], 150 mmol/L NaCl, 3.4 mmol/L EDTA and 0.005% surfactant P20 at pH 7.4) as a running buffer at a constant flow rate of 20 µL/min at 25 °C. The protein was immobilized directly and covalently on the hydrophilic carboxymethylated dextran matrix of the CM5 sensor chip (BIAcore) by using the standard primary amine coupling reaction. The protein to be bound to the sensor chip was diluted in 10 mmol/L sodium acetate buffer (pH 6.5) to a concentration of 17 µmol/L. The concentrations of the compounds dissolved in the running buffer varied from 1.18 to 10 µmol/L. All the data analyses were carried out using BIAevaluation software, and the sensorgrams were processed by automatic correction for nonspecific bulk refractive index effects. The kinetic analyses of the ligand binding to the protein were performed based on the 1:1 Langmuir binding fit model according to the procedures described in the software manual.
Fluorescence titration assay Fluorescence measurements were performed on a Hitachi(Tokyo, Japan) F-2500 fluorescence spectrophotometer equipped with a thermal controller. The change in the intrinsic tryptophan fluorescence when the compound bound to the protein (CypA or CypD) was monitored using a procedure similar to that described in the literature[23,24]. The experiments were carried out at 25 °C in PBS (pH 7.3) with the protein concentration set at 13 µmol/L and the compound concentrations varied from 0 to 40 µmol/L. The compounds were prepared in dimethylsulfoxide as a stock solution of 10 mmol/L. The fluorescent absorption was recorded with excitation at 280 nm and emission at 340 nm.
PPIase inhibition activity assay The PPIase activity assay for the proteins CypA and CypD was performed based on a published method[25] with some modifications. The substrate N-succinyl-Ala-Ala-Pro-Phe-p-nitroanlilide (Suc-AAPF-pNA, S-7388) and α-chymotrypsin (C-7762) were purchased from Sigma (St Louis, Missouri, USA). Suc-AAPF-pNA was dissolved in tetrahydrofuran containing 400 mmol/L of LiCl, and the stock solution concentration was 10 mmol/L. α-chymotrypsin was dissolved in 1 mmol/L HCl containing 2 mmol/L CaCl2, and the stock solution concentration was 80 mmol/L. The assay buffer (173 µL of 50 mmol/L HEPES, 100 mmol/L NaCl; pH 8.0 at 0 °C; final concentration 43 mmol/L HEPES, 86 mmol/L NaCl), 15 µL of de-ionized water and CypD (2 µL of a 2700 nmol/L stock solution) and the compounds (final concentration ranging from 100 nmol/L to 50 µmol/L) were pre-equilibrated for 3 h on ice. Immediately before the assay was started, 7.5 µL of chymotrypsin solution was added. Absorbance readings at 390 nm were recorded when 2.5 µL of the peptide substrate was added into the 1 cm path length cuvette and the solution was mixed rapidly. The data were collected on a Hitachi U2010 spectrophotometer.
Rat liver mitochondrial swelling and Ca2+ uptake/release inhibition assays The mitochondrial swelling and Ca2+ uptake/release inhibition assays were carried out according to published methods[26]. The mitochondria were isolated by differential centrifugation from the livers of adult Wistar rats (180–200 g) after overnight starvation treatment. The rat livers were excised and washed with 0.25 mol/L sucrose. The fat and connective tissue were removed, and the livers were homogenized (1/10, w/v) using buffer A (250 mmol/L mannitol, 0.5 mmol/L EDTA, 5 mmol/L HEPES, 0.1% bovine serum albumin; pH 7.4) on ice. The homogenate was centrifuged at 1000×g for 10 min in a Biofuge Stratos centrifuge (Hereus Company, Hanau, Germany). The sediment was discarded and the supernatant was centrifuged at 1000×g for 10 min twice. The collected supernatant was then further centrifuged at 10 000×g for 15 min. The pellet (mitochondrial fraction) was resuspended in the test buffer (250 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L HEPES; pH 7.4). The total mitochondrial protein was determined by using the Lowry assay using bovine serum albumin as a standard. Rat mitochondria were added to the test buffer to yield a final concentration of 0.5 mg protein per mL[27]. The tested compounds (100 µmol/L) were mixed with mitochondria for 1 h before CaCl2 (200 µmol/L) was added. Mitochondrial swelling was determined by monitoring absorbance at 540 nm using a Hitachi U2010 spectrophotometer and the mitochondrial Ca2+ uptake/release assay was monitored using a Hitachi F-2500 fluorescence spectrophotometer as described previously[26].
Molecular modeling and docking The CypD sequence from Rattus norvegicus was retrieved from GenBank (GenBank protein ID U68544; http://www.ncbi.nlm.nih.gov). The CLUSTAL W program was used to carry out sequence alignment between the sequences of CypD from Rattus norvegicus and human CypA[28]. The sequence similarity identity between CypD and CypA was 63%, and positives were 81%, making the Protein Data Bank (PDB) of human CypA an ideal template for CypD 3-D model building. The 3-D model of the TrpRS was generated based on PDB templates 1AK4[29], 1AWT[30], and 1NMK[31] retrieved from the Protein DataBase by using the MODELLER program[32] encoded in Insight II[33]. MODELLER uses a spatial restraint method to build up 3-D protein models. The structure of each template protein was used to derive spatial restraints expressed as probability density functions for each of the restrained features of the models. The structure with the lowest violation score and lowest energy score was chosen as the candidate. Refinements of the routine in the Homology module of Insight II were used to adjust the positions of the side chains. Finally, the structural models were optimized using Amber force field[34] with the following parameters: a distance-dependent dielectric constant of 4.0, nonbonded cut-off 10 Å, and Kollman-all-atom charges[34]. The structures were first minimized by steepest descent, then by conjugating the gradient method to the energy gradient root-mean-square <0.05 kcal·(mol·Å)-1. Several structural analysis software packages were used to check the structure quality. The Prostat module of Insight II was used to analyze the bonds, angles and torsions. The Profile-3D program[35] was used for checking the structure and sequence compatibility. The 3-D structures of the compounds GW1–7 were constructed from scratch by Sybyl 6.8[36], and optimized to energy convergence with the Tripos force field and MMFF94 charges.
The major residues possibly comprising the binding site of CypD were identified by sequence alignment with human CypA, and the SiteID program encoded in Sybyl 6.8[36]. The surface structure of the binding pocket was constructed by using the MOLCAD module of Sybyl 6.8.
The DOCK suite of programs is designed to find possible orientations of a ligand in a “receptor” site[37]. The orientation of a ligand is evaluated with a shape-scoring function and/or a function approximating the ligand-receptor binding energy. The shape-scoring function is an empirical function resembling the van der Waals’ attractive energy. The ligand-receptor binding energy is taken to be approximately the sum of the van der Waals’ and electrostatic interaction energies. After the initial orientation and scoring evaluation, a grid-based rigid body minimization is carried out for the ligand to locate the nearest local energy minimum within the receptor binding site. The position and conformation of each docked molecule were optimized using the single anchor search and torsion minimization method of DOCK 4.0. Thirty conformations per ligand building a cycle and 50 maximum anchor orientations were used in the anchor-first docking algorithm. All docked configurations were energy minimized using 100 maximum iterations and one minimization cycle.
Results and Discussion
Synthesis of the compounds Generally, the compounds GW1–7 were synthesized as outlined in Scheme 1.
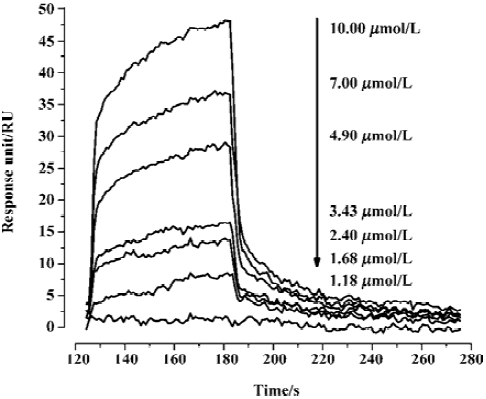
Kinetic analysis of CypD (A) binding to GW1–7 by surface plasmon resonance In order to perform kinetic analyses of the binding of GW1–7 to CypD and CypA, the Biacore 3000 instrument (based on surface plasmon resonance [SPR] technology) was used. As a typical example, the Biacore sensorgrams for the binding of GW2 to the immobilized CypD are shown in Figure 2. The 1:1 Langmuir binding fit model was used for determining the equilibrium dissociation constant (KD), and the association (kon) and dissociation (koff) rate constants by using Equations (1) and (2).
where R represents the response unit, C is the concentration of the analyte, and
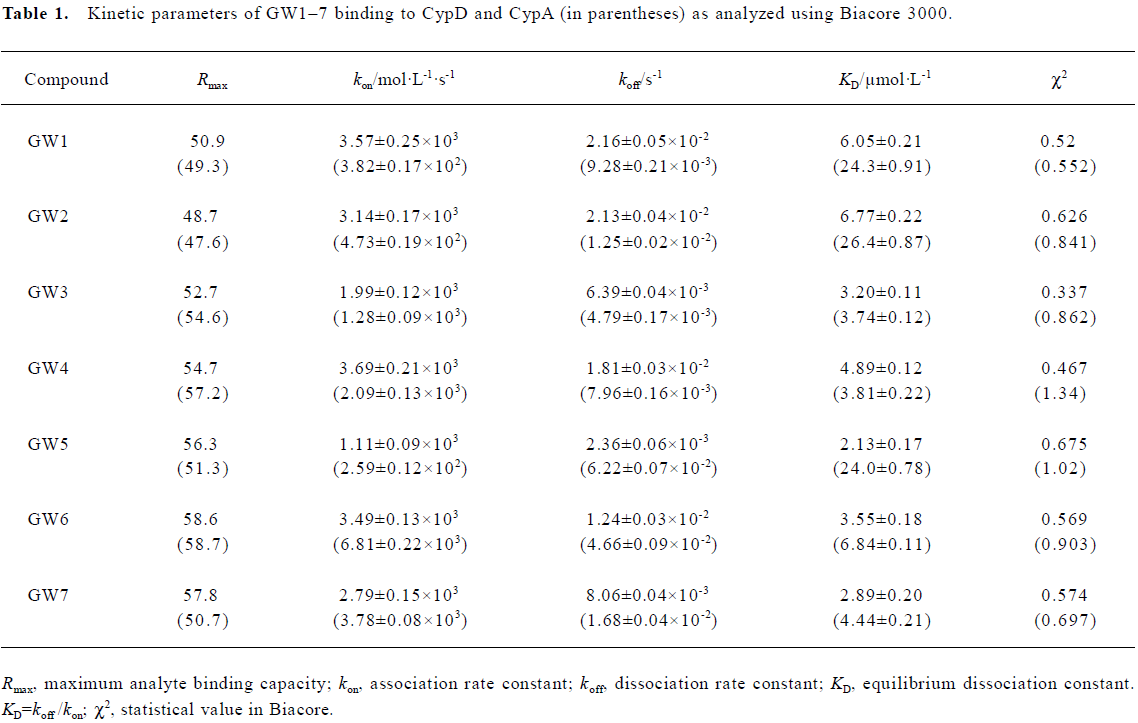
The obtained results were evaluated by χ2 analysis. All the kinetic parameters are listed in Table 1.
Full table
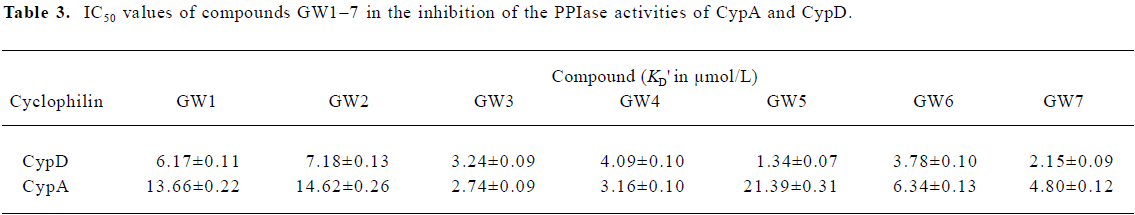
The Biacore results show that all the 7 tested compounds exhibited strong binding affinities with CypD, with KD values approximately 3–6 µmol/L. Due to the high structural homology of CypD and CypA, the tested compounds had high binding affinities with CypA, as indicated in Table 1. However, compound GW5 exhibited higher binding specificity with CypD than with CypA. This was further verified by the intrinsic fluorescence titration analysis and cyclophilin PPIase activity inhibition assay as shown in Tables 2 and 3. Structurally, the R group of the compound (Scheme 1) might play an important role in the ligand binding selectivity for CypD over CypA.
Full table
Full table
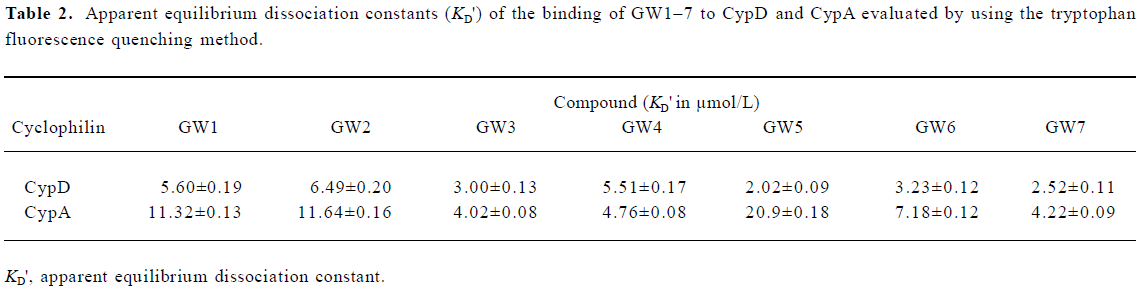
In addition, the KD values obtained from the Biacore assay agreed with the apparent equilibrium dissociation constants (KD') from the intrinsic fluorescence titration analysis and the IC50 values in the cyclophilin PPIase activity inhibition determination as shown in Tables 2 and 3. In agreement with Huber et al[38], we suggest that Biacore is a powerful and useful method for screening cyclophilin inhibitors.
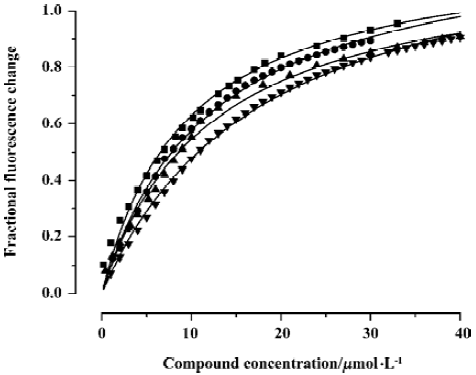
Intrinsic fluorescence titration analysis of compounds binding to CypD(A) Because both of the binding sites of CypD and CypA have a tryptophan residue (Trp 124 for CypD and Trp 121 for CypA), we investigated the binding affinities of the tested compounds for CypD and CypA by using an intrinsic fluorescence titration technique[23]. During the assay, a 1:1 ratio of CypD(A) to binding compound was used based on published information about CypA/CSA interactions[39,40] and the results of our molecular docking analyses. The apparent equilibrium dissociation constant (KD') used for evaluating CypD(A) binding affinity to the tested compound was calculated according to the method in the literature[23]. We assumed that a 50% occupancy of CypD (or CypA) is set at a fractional fluorescence change of 0.5 (FC0.5), and at this point the concentration of the bound ligand is equal to that of the bound protein, which is half of the total concentration of protein. Accordingly, KD' is equal to the total ligand concentration minus the concentration of the bound protein at FC0.5[23].
Figure 3 shows the typical tryptophan fluorescence quenching of CypD induced by titration of the tested compounds with an increase in their concentration. Since none of the compounds showed any intrinsic fluorescence absorption, their possible effects on the experiments could be discounted (data not shown). The KD' values of GW1–7 are summarized in Table 2. Obviously, the binding selectivity of GW5 for CypD over CypA could also be determined, and the KD' values are very comparable to the KD values determined by the Biacore assay.
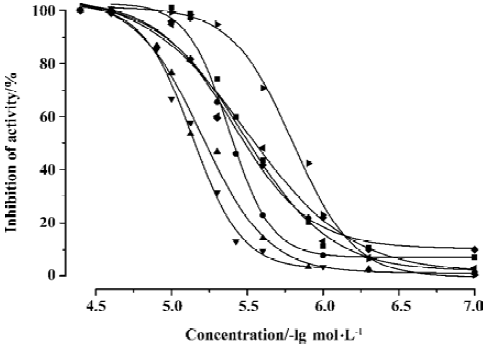
CypD (A) PPIase activity inhibition assay Both CypD and CypA belong to the PPIase family, and classic spectrophotometric methods[41] can be used to determine the PPIase inhibition activity of GW1–7 against CypD and CypA. During the assay, the rate constants for the cis-trans interconver-sion were evaluated by fitting the data to the integrated first-order rate equation by nonlinear least-square analysis[25,41].
As a typical example, Figure 4 shows the CypD PPIase inhibition results with increases in the concentrations of the compounds, and Table 3 shows the IC50 values of GW1–7 against CypD and CypA. The fact that the IC50 values accord well with the Biacore and fluorescence titration results (Tables 1, 2) confirms the reliability of these three detection approaches.
Rat Ca2+-dependent mitochondrial swelling and Ca2+ uptake/release inhibition assays In general, MPT pores are open when mitochondria encounter abnormally high concentrations of exogenous Ca2+ ions. These pores allow solutes of <1500 Da in size across the inner mitochondrial membrane, leading to mitochondrial swelling. Such swelling can be detected by time scans of absorbance at 540 nm (A540) and the extent of swelling is proportional to A540[26].
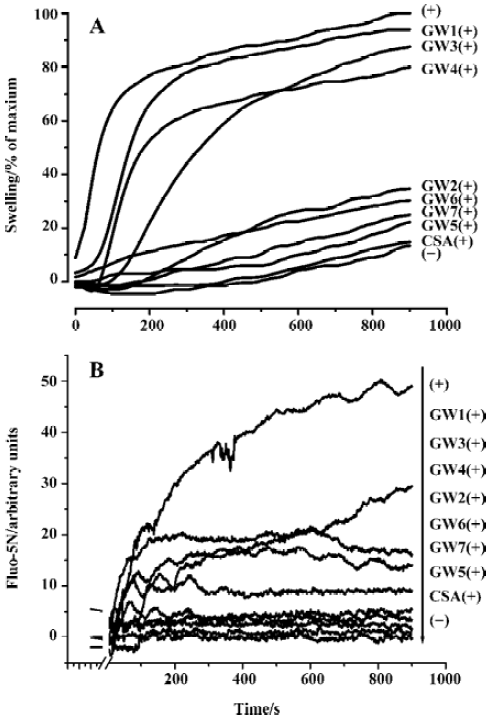
Fluo-5N fluorescence is quite low without binding to Ca2+ in controls, because the high mitochondrial membrane potential prevents the release of endogenous mitochondrial Ca2+. When exogenous Ca2+ was added, Fluo-5N fluorescence increased immediately and decreased rapidly as Ca2+ ions were taken up into the mitochondria. Subsequently, the accumulation of cations in the mitochondria led to mitochondrial swelling and depolarization. Ca2+ ions were then released from mitochondria as a consequence of the onset of MPT, as indicated by an increase in Fluo-5N fluorescence. Ca2+ release was completely blocked by 1 µmol/L CSA. Fluo-5N fluorescence also revealed that compounds GW1–7 inhibited the uptake/release of exogenously added Ca2+ to a certain extent.
Figure 5A shows the results of the rat mitochondrial swelling inhibition assay for GW1–7 (100 µmol/L) with CSA (1 µmol/L) as a control. Figure 5B gives the results of the mitochondrial Ca2+ uptake/release inhibition assay for GW1–7 (100 µmol/L). The results show that the inhibition abilities of compounds GW1–7 against Ca2+ uptake/release are in good agreement with their inhibition abilities against mitochondrial swelling. We found that compounds GW2, 5, 6, and 7 had a strong ability, whereas GW1, 3, and 4 did not have any inhibition activity, which could be because of the R group. Compared with GW2, 5, 6, and 7, the tails of GW1, 3, and 4 were ethoxycarbonyl, which might prevent them from transferring into mitochondria through the membranes or cause the loss of inhibition ability for other (unclear) reasons. The behavior of CypD in mitochondria is much more complicated than that of the purified CypD protein, so the different R groups might cause different results. CSA had the highest inhibition activity, and the relative general inhibition abilities of the other compounds were: GW5>GW7>GW6>GW2. Such a sequence seems to be consistent with the CypD PPIase inhibition ability of the compounds (Table 3). This result thus confirms the fact that CypD inhibitors may possess possible inhibition activity against Ca2+-dependent MPT pore opening.
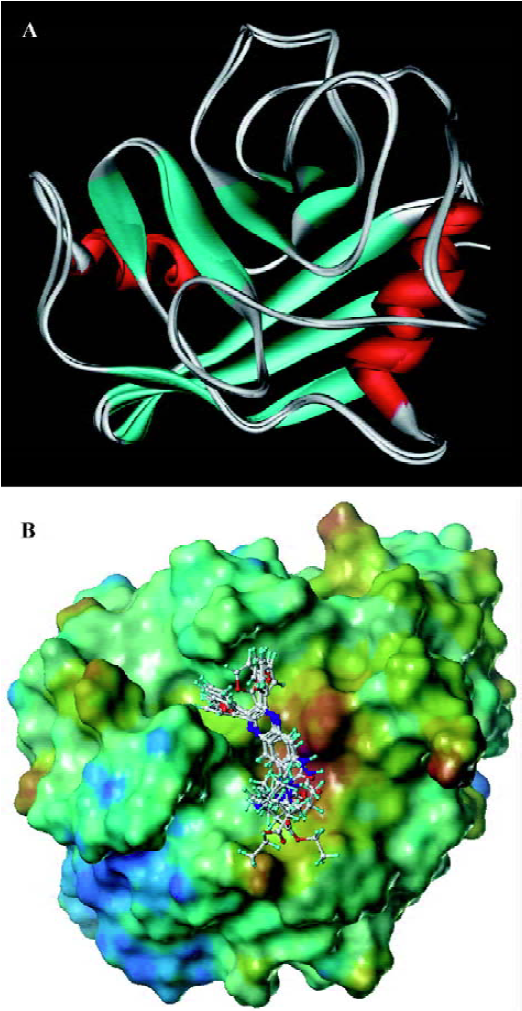
Molecular docking analyses To gain further insight into the CypD(A)/inhibitor interaction model at the atomic level, docking analysis based on molecular modeling was carried out without the published rat CypD crystal data. Our rat CypD model tallies very well with the human CypD crystal[PDB ID: 2bit] structure shown in Figure 6A. The weighted root mean square distance is 0.6040 and the identity score is 95.7%. Because of the similar structures of the compounds, they share the same precursor, with some overlapping structural elements in common (Figure 6B). Similar to CypA, the binding pocket of CypD is also fairly large and shallow, and is composed of residues Arg58, Ile60, Phe63, Met64, Glu66, Gly75, Thr76, Gly77, Ala104, Asn105, Ala106, Gln114, Phe116, Thr122, Trp124, Leu125, Lys128, and His129. GW1–7 and CSA bind to the same binding site of CypD. Unlike the case of full occupation by CSA, GW1–7 occupied only part of the binding pocket and might swing in the pocket. Helekar and Patrick even demonstrated that Arg55 of CypA was a key determinant against PPIase activity[42]. Compounds GW1–7 showed their hydrophobic contact with Arg58 of CypD (Arg55 of CypA). Therefore, the PPIase activity of CypD could be inhibited by hydrophobic interactions with the inhibitors. In addition, GW1–7 formed stacking interactions with Trp124 of CypD (Trp121 of CypA) and the only tryptophan residues of CypD and CypA that contribute to the change in fluorescence intensity (data not shown).
Conclusion In this work, we reported on 7 small quinoxa-line derivatives as novel CypD inhibitors. In vitro assays indicated that compounds GW2, 5, 6, and 7 inhibit Ca2+-dependent rat liver mitochondrial swelling and Ca2+ uptake/release. By using SPR and fluorescence titration techniques, kinetic analysis of CypD/inhibitor interactions were quantitatively performed. The measured IC50 values for the tested compounds are all in good agreement with the SPR and fluorescence titration results, which suggests that these are powerful methods for identifying CypD inhibitors[38]. Further studies indicated that GW5 has binding selectivity for CypD over CypA.
In summary, in this present work we used an appropriate and powerful approach for identifying CypD inhibitors, and developed a small compound that shows specific ligand-binding ability for CypD, which could be used in the inhibition of MPT pore opening.
Acknowledgements
We thank Dr James D LECHLEITER for providing the rat CypD gene and Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) for providing human cyclophilin D structure information.
References
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996;86:147-57.
- Susin SA, Lorenzon HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999;397:441-6.
- Olson M, Kornbluth S. Mitochondria in apoptosis and human disease. Curr Mol Med 2001;1:91-122.
- Crompton M. Mitochondria and aging: a role for the permeability transition? Aging Cell 2004;3:3-6.
- Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem 2003;10:1485-506.
- Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med 2003;9:196-205.
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 2005;79:231-9.
- Crompton M. On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr Med Chem 2003;10:1473-84.
- Connern CP, Halestrap AP. Purification and N-terminal sequencing of peptidyl-prolyl cis-trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem J 1992;284:381-5.
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989;337:476-8.
- Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J Biol Chem 2002;277:31134-41.
- Li Y, Johnson N, Capano M, Edwards M, Crompton M. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem J 2004;383:101-9.
- Schubert A, Grimm S. Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res 2004;64:85-93.
- Machida K, Osada H. Molecular interaction between cyclophilin D and adenine nucleotide translocase in cytochrome c release: does it determine whether cytochrome c release is dependent on permeability transition or not? Ann NY Acad Sci 2003;1010:182-5.
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341:233-49.
- Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie 2002;84:153-66.
- Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur J Biochem 1988;178:489-501.
- Crompton M, Barksby E, Johnson N, Capono M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 2002;84:143-52.
- Alaimo RJ [inventor]. Norwich Eaton Pharmaceuticals [assignee]. Thiocyanatoquinoxaline compounds with immunomodulating activity. US patent 4540693. September 10, 1985.
- Magnus P, Thurston LS. Synthesis of the vinblastine-like antitumor bis-indole alkaloid navelbine analogue desethyldihydronavel-bine. J Org Chem 1991;56:1166-70.
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 1989.
- Luo C, Luo H, Zheng S, Gui C, Yue L, Yu C, et al. Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A. Biochem Biophys Res Commun 2004;321:557-65.
- Husi H, Zurini MGM. Comparative binding studies of cyclophilins to cyclosporin A and derivatives by fluorescence measurement. Anal Biochem 1994;222:251-5.
- Handschumacher RE, Harding MW, Rice J, Drugge RJ. Cyclophilin A: a specific cytosolic binding protein for cyclosporin A. Science 1984;226:544-7.
- Kofron JL, Kuzmic P, Kishore V, Colon-Bonilla E, Rich DH. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 1991;30:6127-34.
- Blattner JR, He L, Lemasters JJ. Screening assays for the mitochondrial permeability transition using a fluorescence multiwell plate reader. Anal Biochem 2001;295:220-6.
- Lowry OH, Rosenbrough NH, Farr AL, Randall JR. Protein measurement with the folin phenol reagent. J Biol Chem 1951;193:265-75.
- Thompson J, Higgins D, Gibson T. CLUSTAL_W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673-80.
- Gamble T, Vajdos F, Yoo S, Worthylake D, Houseweart M, Sundquist WI, et al. Crystal structure of human cyclophilin a bound to the amino-terminal domain of HIV-1 capsid. Cell 1996;87:1285-94.
- Vajdos F, Yoo S, Houseweart M, Sundquist W, Hill C. Crystal structure of cyclophilin a complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci 1997;6:2297-307.
- Sedrani R, Kallen J, Martin Cabrejas L, Papageorious C, Senia F, Rohrbach S, et al. Sanglifehrin-cyclophilin interaction: degradation work, synthetic macrocyclic analogues, x-ray crystal structure and binding data. J Am Chem Soc 2003;125:3849-59.
- Sali A, Blundell T. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779-815.
- Insight II [molecular modeling package]. San Diego, California, the United States: Molecular Simulations; 2000.
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, et al. A Second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 1995;117:5179-97.
- Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991;253:164-70.
- Sybyl [molecular modeling package]. St Louis, MO: Tripos Associates; 2000.
- Ewing T, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem 1997;18:1175-89.
- Huber W, Persicace S, Kohler J, Muller F, Schlatter D. SPR-based interaction studies with small molecular weight ligands using hAGT fusion proteins. Anal Biochem 2004;333:280-8.
- Kallen J, Spitzfaden C, Zurini MGM, Wider G, Widmer H, Wuthrich K, et al. Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature 1991;353:276-9.
- Pfluefel G, Kallen J, Schirmer T, Jansonius JN, Zurini MGM, Walkinshaw MD, et al. X-ray structure of a decameric cyclophilin-cyclosporin crystal complex. Nature 1993;361:9-4.
- Fischer G, Berger E, Bang H. Kinetic β-deuterium isotope effects suggest a covalent mechanism for the protein folding enzyme peptidylprolyl cis/trans-isomerase. FEBS Lett 1989;250:267-70.
- Helekar S, Patrick J. Peptidyl prolyl cis-trans isomerase activity of cyclophilin A in functional homo-oligomeric receptor expression. Proc Natl Acad Sci USA 1997;94:5432-7.