
Introduction to 3D reconstruction of macromolecules using single particle eleActron microscopy1
Introduction
Transmission electron microscopy (TEM) is a structural technique that has existed for many years in biology to study the ultra-structure of cells. However, it has only been more recently that outstanding technical advances have consolidated the prospect of observing single protein molecules at the electron microscope level with such sufficient structural details to use these data to reconstruct the molecule in three dimensions (3D). These methodological improvements lie at the level of the instrument itself, that is, better microscopes, but most significantly, more powerful algorithms and software platforms, and, importantly, dramatically increased speed of computers to deal with the noisy images of proteins obtained with the microscope. As a result of these advances, analysis of macromolecules using single-particle electron microscopy (EM) can be widely noticed in a fast-increasing number of publications. Hence, the need rapidly emerged to store and make accessible all of this 3D information to the scientific community, and the Macromolecular Structure Data Base (at the European Bioinformatics Institute, Cambridge, UK) has been created to this end (http://www.ebi.ac.uk/msd/). Most scientific journals now make mandatory the deposition of any 3D structure obtained by EM into this data base, importantly using standard formats to guarantee the interchange of the data. This will certainly stimulate both the flow of EM structures among scientists (as already happens with atomic coordinates) as well as the quality of the EM work deposited.
As EM spreads out, its interaction and dependence on other structural techniques has deepened. A modern approach to the exploration of macromolecular structures requires a wise combination of molecular biology, biochemistry, biocomputing, X-ray crystallography, nuclear magnetic resonance (NMR), EM and any other structural technique (ultracentrifugation, small-angle neutron scattering, etc). An important challenge will therefore be to develop methods to combine all of this multi-resolution information in a comprehensive way.
In the following sections, I will describe the major methods that EM employs today, how relevant structural information is extracted, and the present limitations of these approaches. Supplementary and more in-depth information can be found elsewhere[1–9] and on the web (eg the 3D-electron microscopy data base at http://3dem.ucsd.edu/index.html, the Electron Microscopy Yellow Pages at http://cimewww.epfl.ch/EMYP/comp.html, or the SPIDER web site at http://www.wadsworth.org/spider_doc/spider/docs/spider.html).
Basics of single-particle electron microscopy
The protein of interest must be purified to homogeneity prior to any EM analysis, as image processing is most generally based on the assumption that every single image we take derives from the same specimen (Figure 1A). The sample is then applied to EM grids covered by a thin carbon support film (which can contain small holes in the case of cryo-EM, where specimens are vitrified) and visualized under the electron microscope (Figure 1B). Molecules of the protein adsorb to the carbon film in orientations determined by the charges on their surface and their overall shape. Ideally, a random distribution of orientations is desirable, because this will allow recording images of the protein from many different angles, a requirement to obtain a correct 3D reconstruction. Before insertion into the microscope, the sample must be prepared to withstand the incident radiation (electrons). For this purpose, the protein on the EM grid can be either stained with heavy-atom salts, known as negative staining (uranyl acetate, uranyl formiate and ammonium molibdate, as the most widely used staining agents), or quickly vitrified into liquid ethane and kept under liquid nitrogen temperatures (cryo-EM). Each method has its own advantages and limitations widely discussed before and beyond the scope of this review[5,7,10]. Briefly, negative staining provides a higher contrast at the expense of resolution and only the surface or topography of the molecule is actually defined. Performing cryo-EM experiments is technically more demanding for the microscopist, but perfectly preserves the structure of the protein at high resolution within the vitrified buffer. Nevertheless, contrast is strongly reduced, thus small proteins might only be analyzed with the help of staining agents[10–12].
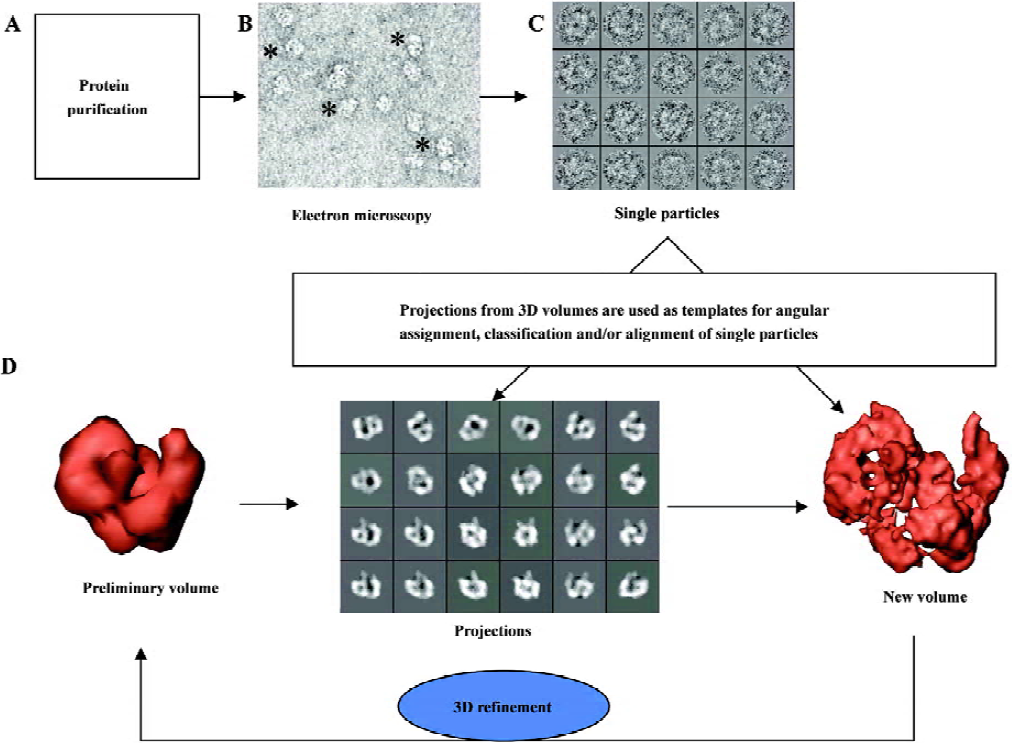
Images obtained with the electron microscope are projections of the molecules along the direction of the electron beam. In a very simplified view, we can state that as the beam encounters more atoms along its path within the protein, the fewer electrons get into the detector, either photographic film or CCD, therefore integrating the 3D information of the molecule along the beam direction. Large collections of images from single molecules are selected and boxed out from each micrograph (after digitalization) or CCD frames (Figure 1C), which become the starting data set for image processing. These single particle images are always noisy, because low levels of electrons are used during imaging to reduce radiation damage and so minimize the destruction of the structural information. Furthermore, single particle images are intentionally under focus to secure sufficient contrast of the protein over the background, so molecules can be identified within the micrographs. Both the high noise levels and the under focus of the micrographs are responsible for the experimental limitations to reach high resolution in 3D reconstructions using EM. Consequently, the aims of image processing (Figure 1D) are 2-fold: first, to reduce the noise present in the images by averaging similar projections in 2 dimensions and, later on, into a 3D volume; second, to correct the consequences of under focus and other optical effects during the generation of images in the microscope (globally known as contrast transfer function, CTF). This second aspect of image processing will not be discussed in this review, but it has been nicely introduced elsewhere[7,13,14]. Generally speaking, image processing in 2D is required at some stage in order to classify those images corresponding to similar projections of the molecules (therefore, similar “shape”) and to align them in 2D, this is, to place them into register, so that they can be averaged pixel-by-pixel to improve their signal-to-noise ratio. How is a 3D structure then reconstructed from the 2D data recorded? It is demonstrated that 2D projections along a 3D object contain sufficient information to restore the original object if the orientation angles of each projection are known, and several algorithms and approximations can easily perform this task. For instance, in medical tomography a radiation source is used to acquire projections of the patient along a set of established directions and then a 3D reconstruction is generated. Just a word of caution to point out that several algorithms exist to reconstruct a 3D structure from its projections at known orientations and the mathematics behind them and its relevance to the correctness of the resulting structure is not insignificant[15–17]. In single-particle EM, different views of the same protein are contained within each micrograph as, frequently, molecules interact with the support film or are enclosed within the vitrified ice (in cryo-EM) at many different orientations. In order to generate a “correct” 3D structure, all these projections of the molecule must evenly fill Fourier space, meaning we are merging in 3D images from all possible angles. Nevertheless, there are cases where the shape of the molecule can make it mathematically redundant to collect all possible views. For instance, GroEL, a molecular chaperon made up of 2 back-to-back stacked rings can be reconstructed just from its side views because the protein rotates along its longitudinal axis filling all Fourier space without the need to incorporate top views during image processing[18,19]. Therefore, once a sufficient data set is collected, the only requirement to resolve the 3D structure of the protein is to establish the orientation of each projection image with respect to a common set of reference coordinates. The problem of reconstructing a volume from projections is mostly reduced to that of angular assignment. This is actually the most time- and effort-consuming task during single particle EM and the heart of the image processing itself (Figure 1D). Several software platforms are commonly used, each one with a specific vision on how to approach the problem of angular assignment and 3D reconstruction, which in all cases incorporates some type of iterative refinement of the data (Figure 1D).
These procedures described above require images from the same molecule at the same conformation taken in several orientations. In some cases, such data cannot be collected either because the protein binds to the grid in a preferred view, or because conformational flexibility exists in the protein, and therefore different views cannot be unambiguously assigned to a specific conformation. In such situations, the random conical tilt method can deliver a 3D structure for each type of view and generate a volume without a template[20–22].
Several commonly used software packages can do the job
Along the already relatively extended history of electron microscopy and image processing, several groups have deposited a lot of effort into the development of theory, methodological approaches, algorithms and complete platforms for the analysis of single-molecule images taken under the electron microscope. It is an outstanding effort that all microscopists should thank because they provide us with the tools we need in our everyday work. Original work by Crowther and colleagues developed the first methods to combine images of the same specimen lying at different orientations and applied them to icosahedral viruses[23,24]. Moreover, works performed on the structural determination of viral capsids have led the way in the possibilities of 3D EM. Accordingly, in 1997 2 groups, led by Crowther at Cambridge (UK) and Steven at the National Institutes of Health in Bethesda (USA), managed to visualize secondary structural elements in the 3D structure of hepatitis B viral cores at 7.4 Å and 9 Å[25,26], a major breakthrough at the time and the beginning of today’s improvements in the EM field.
At present, some kind of implicit standard has been reached and most EM work is performed using 1 of 3 distinct software platforms: SPIDER[27], IMAGIC[28] and EMAN[16]. As a note of prudence, other popular platforms and programs exist, many of them dedicated to the processing of particles with icosahedral symmetry[29], which will not be discussed in this review. SPIDER was developed by the group of Joachim Frank in Albany (NY, USA), IMAGIC by Marin van Heel’s group in London (UK) and EMAN by Steve Ludtke and Wah Chiu in Houston (TX, USA). EMAN is the most recent platform and it is available completely free of charge. More information about each package is found on their respective web sites: SPIDER (http://www.wadsworth.org/spider_doc/spider/docs/spider.html), IMAGIC (http://www.imagescience.de/imagic/) and EMAN (http://ncmi.bcm.tmc.edu/ncmi/).
Other research groups are also intensively contributing to software development and implementation for EM analysis, whose strength lies in that they mostly attend to aspects or approaches in image processing not sufficiently looked after by the previous platforms. This software can therefore add force to the potential of the most commonly used packages. This is the case with XMIPP, which includes a good repertoire of classification and alignment algorithms[30–32] (http://www.cnb.uam.es/~bioinfo/), FREALIGN (http://emlab.rose2.brandeis.edu/grigorieff/downloads.html) designed for extracting high-resolution features at the final stages of refinement, and BSOFT (http://www.niams.nih.gov/labbranch/lsbr/software/bsoft) containing, among other tools, a good algorithm to estimate and correct the CTF of the micrographs. To this day, the most common way for EM groups to make use of all of these computational possibilities is to choose 1 or 2 of the above main platforms while using other software to complement them for specific tasks.
It is as well worth mentioning that these efforts in software development for EM processing have been matched by spectacular improvements in the programs needed to render and visualize the 3D data. Many different programs are now available, all of them very good, each displaying advantages in specific features. Just a few of the most typically used by the EM community are AMIRA (http://www.amiravis.com/), CHIMERA (http://www.cgl.ucsf.edu/chimera/), VMD (visual molecular dynamics; REF; http://www.ks.uiuc.edu/Research/vmd/) or PYMOL (http://pymol.sourceforge.net/).
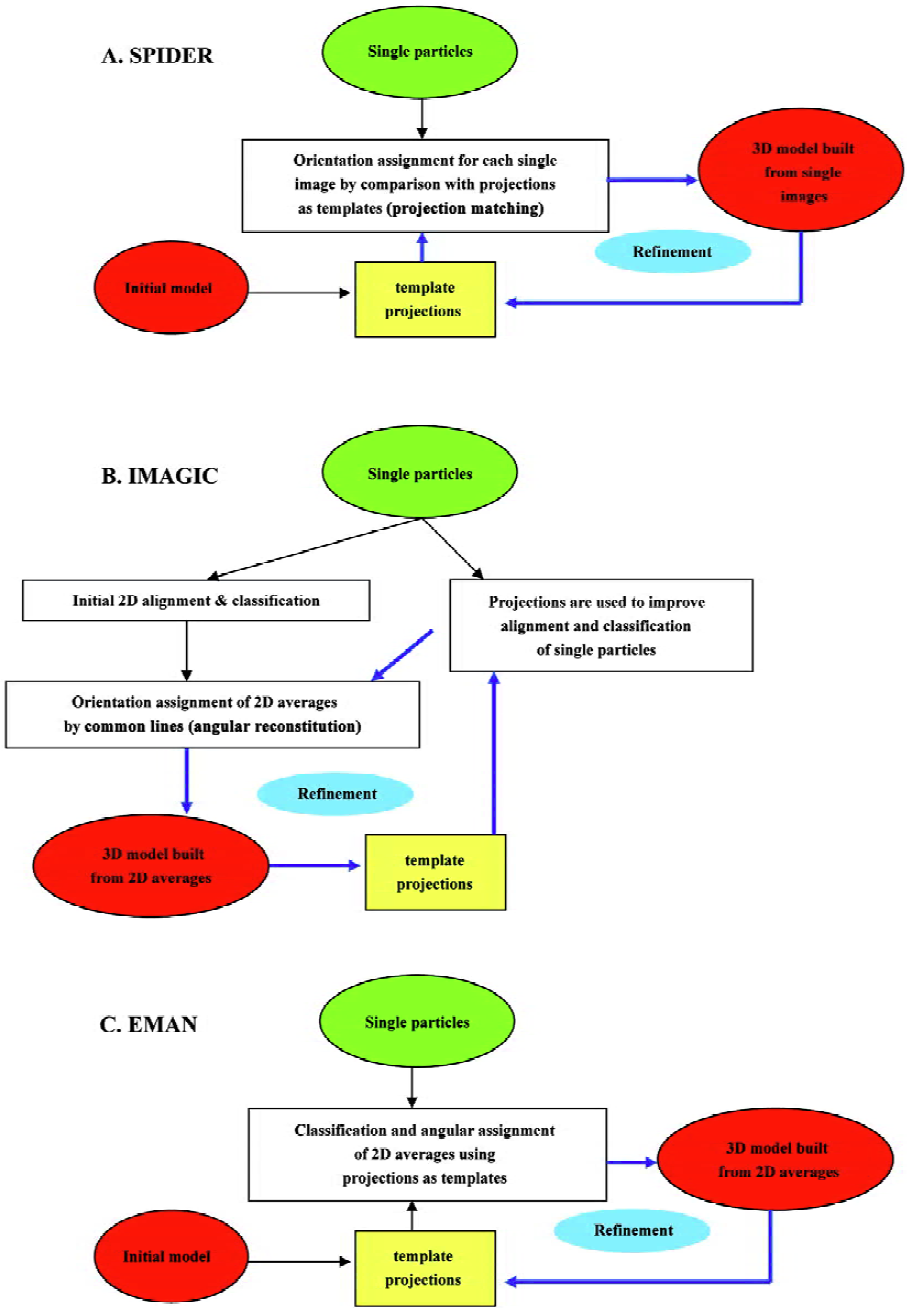
In the following paragraphs I will summarize the basic features of each one of the main platforms (SPIDER, IMAGIC and EMAN)[16,27,28], pointing out those aspects that make each software package exceptional (Figure 2). Globally, the main differences among them center on (i) the use of either single particles or their 2D averages to build the volumes; and (ii) the means for angular assignment, either “angular reconstitution” or “projection matching”[7]. With respect to the first point, all 3 platforms use the images from single particles as input data, but only SPIDER directly utilizes these to reconstruct the volume, because both IMAGIC and EMAN classify and average single images from similar views of the protein in order to produce a 2D average with improved signal-to-noise ratio. These averages then constitute the input to reconstruct the 3D structure. With respect to the second point, and as mentioned earlier, the fundamental aspect of image processing corresponds to the determination of the orientation angle of each image (or average) with respect to a common set of coordinates. SPIDER and EMAN define these angles by comparison with projections of preliminary volumes that act as templates of known angles. Within each cycle of refinement the reconstructed volumes and their projections are improved, so that angular assignment is also iteratively improved. This strategy is known as “projection matching”. Alternatively, IMAGIC defines orientation angles using “common lines”, an algorithm that can potentially find the angular relation between projections without additional input. I will not get into the principles that underlie common lines but this requires a high signal-to-noise ratio to diminish false solutions and, consequently, IMAGIC spends much of its efforts in particle classification, alignment and averaging. Its great conceptual advantage is that angles come directly from the data, thus the name “angular reconstitution”[7]. Model bias in the assignment of angles is therefore greatly reduced, though some bias still exists because projections from iteratively improved 3D models are used to increase the accuracy in particle alignment and classification. It is imperative to point out that, besides these differences in the general approaches among several platforms, each of these contains the tools required to perform almost any operation with the images from the electron microscope, and are consequently intrinsically very flexible. Therefore, for instance, a “projection matching” strategy can be perfectly carried out using tools provided by IMAGIC.
Figure 2 outlines a generalised flow-through during image processing with each platform. SPIDER[27] initiates from a rough starting model to generate projections of defined angular spacing (Figure 2A). Each single particle is compared with all projections so that it receives those angles of that template with which it better fits. This preliminary angular assignment is used to build a new 3D model that acts as a new source for projections. As this process is repeated iteratively (“angular refinement”), projections better match the real data and, at the end, the angles assigned to the particles allow reconstruction of the structure. Refinement in IMAGIC[28] (Figure 2B), on the other hand, makes use of the projection templates just to align the particles, so classification and averaging can be iteratively improved, but angles are defined using the 2D averages and common lines. Finally, EMAN[16] (Figure 2C) has adopted a scheme somehow in between those of SPIDER and IMAGIC. Angular assignment for each particle in EMAN is defined based in their correlation with projection templates, as with SPIDER. But instead of using particles directly to build the volumes, all particles with a similar orientation constitute a group or class to be averaged, and only these averages are then used to reconstruct a volume. The process of averaging in EMAN incorporates a very good set of parameters that can be tuned to improve averaging and discard “bad” particles. An especially interesting feature of EMAN is that particles within a class are actually “refined” during averaging so that model bias is strongly minimised and single images with a standard deviation above a certain threshold are not incorporated into the final average. Common to all 3 systems is that either mechanism of angular assignment is repeated iteratively (angular refinement) until the angles assigned to the particles and the resulting 3D structure are stabilised. At the end, if correctly used, any of these 3 software platforms can construct an accurate structure. Nevertheless, it is extremely important to note that image processing is far from a fully automated method that does not require user intervention. On the contrary, each processing platform just provides a large number of computing tools to deal with the data from the microscope, but evaluation of the output results and decisions during processing are completely user dependent. Consequently, an inexperienced user could end up with a wrong structure.
The resolution problem or how to solve the resolution gap
Once we have the final 3D reconstruction of our macromolecule, the last stage of the research involves the in-depth inspection and description of the structure. This is a crucial step because interpretation of the 3D data is the source for the extraction of biologically relevant information, and therefore the source of our conclusions about the processes we are studying. In single-particle EM this task is problematic because the structures are solved to resolutions above those required to trace the polypeptide chain, due to the difficulties still present during averaging and alignment of the noisy images obtained with the microscope. Typically, EM analysis provides structures ranging from 8 Å–10 Å to 30 Å–40 Å resolution, and the consequences of these resolutions for the way in which a macromolecule is visualized can be perceived in Figure 3C, where I have used as an example the recent atomic model of DNA-PKcs, a kinase implicated in DNA repair[33]. While at a resolution of 9 Å secondary structural elements, such as alpha helices, can still be distinguished in favorable cases[19,34], at poorer resolutions (>15 Å), rarely anything more than the overall shape of the protein is apparent (Figure 3C).
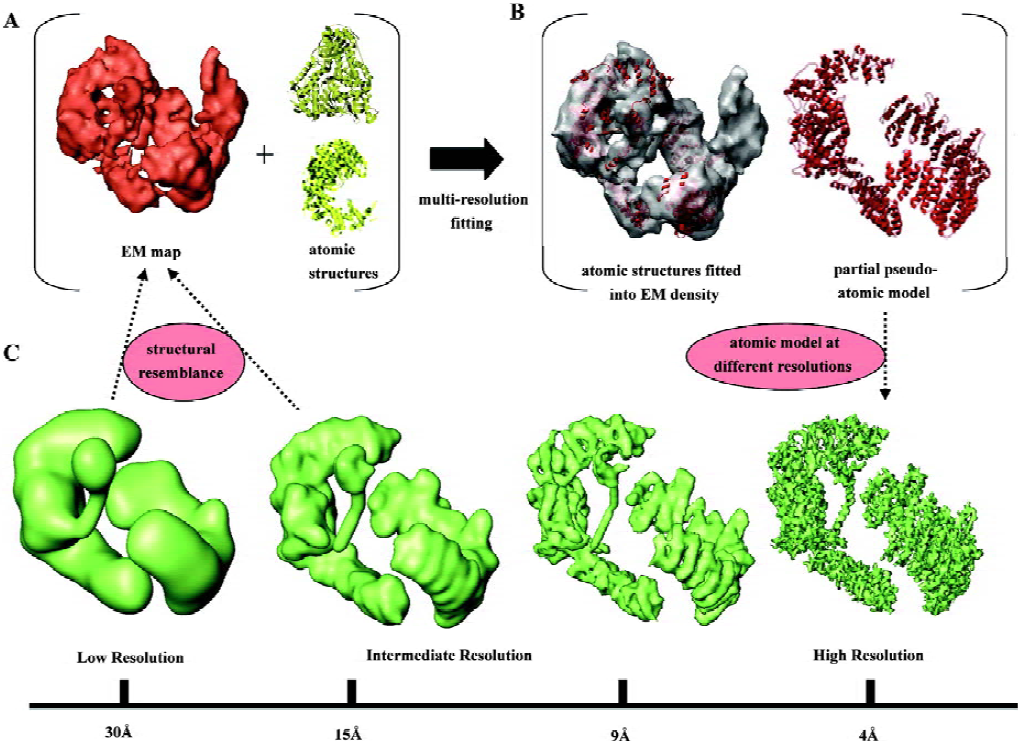
To bridge the gap between the atomic information we would wish to have in our structure and the medium or low resolution of the actual EM reconstructions, 3 “multi-resolution” methods have been proposed[35–39]. These suggest combining information at different levels of resolution in order to investigate complex systems.
Rigid-body fitting A medium-resolution EM structure can be depicted as a convolution of its atomic features; hence a pseudo-atomic model can be obtained by computationally placing (“fitting”) atomic coordinates into the EM map[36]. The atomic structure is considered to be a rigid body with no conformational changes, whose density has to be located and placed within the 3D reconstruction. Rigid-body fitting is a very appropriate mode to map domains into a larger complex that contains several domains or proteins, and it has been extensively used already to propose pseudo-atomic models of macromolecular complexes. However, it is important to bear in mind that the accuracy of this computational approach can be seriously hampered by a lack of resolution of the target 3D model and by the size, shape and conformational flexibility of the fitted atomic structure. Figure 3 shows a recent example from our group where the 3D structure of the DNA-PKcs kinase[33] was fitted with atomic structures of individual domains (Figure 3A) to produce a pseudo-atomic model for some portions of the molecule (Figure 3B). This atomic model, when filtered to the resolution experimentally obtained by EM, very much resembles the corresponding segments of the 3D structure (Figure 3C). Many other examples can be found in the recent literature where these methods have been applied[40–42].
Several algorithms have been developed that consider different sides of the problem[35,36], but this is still a very active field where a consensus about the best approaches has not yet been reached. Some of the most commonly used tools are those implemented in SITUS[43] (http://situs.biomachina.org/), EMAN[16] (http://ncmi.bcm.tmc.edu/ncmi/) and EM fit[44]. Nevertheless, other algorithms are also frequently used[45–47]. At low resolution, several solutions could comply with our fitting criteria, so one must be cautious with the results, which, if much uncertainty persists, should ideally be supported or validated with external information.
Flexible fitting It is very likely that an atomic structure will not perfectly match one solved by EM, especially when the portion represented by the atomic structure is part of a larger complex solved by EM or it is at a different stage of its functional cycle. In such cases, and if sufficiently good resolution is present, the atomic coordinates can be modified so that they better fit into the EM density. By doing so, we can not only place a domain within a larger structure (as in rigid-body fitting), but we can also actually identify a new conformation of the protein. A nice example has been shown recently in the 6 Å structure of GroEL, where some displacement of helices were found compared to its atomic coordinates[19]. Both SITUS[48] and EMAN[16] contain algorithms to perform flexible fitting.
Prediction of secondary structure elements and protein folds In those cases where the resolution of an EM map is very good (usually anything below 8 Å), instead of fitting known atomic structures, a more powerful approach can be carried out, and the actual recognition of secondary structure elements within the map can be achieved. The AIRS platform in EMAN has several commands (helixhunter, ssehunter) to look for sheets and helices in the maps. A score is supplied to help discriminate real from spurious findings. In favorable cases, a whole fold type can be defined and consequently a sufficiently realistic atomic model. A brilliant example has been recently published describing a pseudo-atomic model for the capsid of phi29 phage[34]. Other groups are also currently working on fold predictions from EM reconstructions[49]. Importantly, results in the area of secondary structure prediction need to be handled with great care due to the still innovative nature of this field.
Some notable recent electron microscopy studies
The last 2–3 years have been characterized by a rapid increase in EM publications. I wish to point out some significant works recently published, which have been dealing with some challenging applications of single-particle EM. These works shed light on where the field is going in the very near future. I strongly apologize to all whose work has not been reflected in this review.
Small, asymmetric and flexible proteins Certainly, EM has clear limitations with respect to the smallest size of the molecules it can analyze. Proteins must be visualized above a noisy background to be extracted and processed, and only large macromolecules can therefore fit this criterion. There are also deeper conceptual reasons that do not allow reconstruction of very small molecules[50]. As a result, proteins targeted by EM studies are usually above 200 kDa–300 kDa. Having said that, some recent outstanding works have challenged these difficulties and studied small proteins, many with molecular weights around, or even below, 100 kDa, and these have been reconstructed at decent resolutions (around 20 Å–30 Å): geminin[51], separase[11], the Arp2/3 complex[52] and the mammalian fatty acid synthase[53], to name a few.
A common feature of many of these proteins is that they participate in very relevant pathways in the biology of the cell (eg signaling pathways, DNA repair, oncogenes and tumor suppressors), and though they are not extremely large, they are still difficult to purify in large quantities for X-ray studies. This is the case, for instance, with Vav, an activator of Rho/Rac GTPases, whose 3D structure has been solved in the inactive and active conformations, plus a ~85 kDa truncated mutant with oncogenic potential[12], providing insights into its regulatory mechanisms. Another remarkable example is the structure of the tetrameric KvAP voltage-dependent K+ channel[54] with a mass of 100 kDa, which the authors deliberately increased up to 300 kDa by addition of 4 Fab fragments.
Generally speaking, all of these works have the challenging difficulty of collecting good microscopy data and being able to correctly align images of small molecules, which, on top of that, frequently display no symmetry at all. But an even further twist to these already difficult experiments comes when the protein under study is flexible; several conformations are present in the same micrographs whose identification is not always straightforward. In some of these cases, the method called “random conical tilt”[20,22] (developed some time ago to obtain 3D structures from proteins bound to the EM grids with preferential views) might be the only tactic available to solve these structures without mixing several conformations. In this regard, it is worth taking a look at some recent excellent works by Tomas Waltz’s group[55,56]
Structures of the ribosome The complex structure of the ribosome and the process of mRNA translation have been extensively studied by several groups in the last decade. These authors regularly provide structures at resolutions better than 12 Å and the wealth of biological information obtained by combining EM and X-ray crystallography of the ribosome is unprecedented. Two groups have been leading this research: Joachim Frank at Albany (NY, USA)[57–61] and Marin van Heel at London (UK)[62–64].
Multi-protein macromolecular complexes or “molecular machines” The natural targets of EM are those complexes that contain several proteins, are very large in size, are heterogeneous in their composition, have complex functional cycles and are difficult to purify; all of these qualities making complicated a traditional analysis by crystallography or NMR. Interestingly, these large and transient complexes, sometimes named “molecular machines”, have been admitted by modern biology to comprise the basics for a major number of cellular processes, and are therefore a subject of great interest. Some EM works have started to obtain 3D information on some of these complexes, such us the spliceosome (implicated in mRNA splicing)[65–68], the SAGA complex from Saccharomyces cerevisiae[69], the apoptosome involved in procaspase-9 binding and activation during apoptosis[70], and several complexes between chaperonins with their substrates and co-chaperons[71–74].
DNA-bound protein complexes Determination of the 3D structure of complexes between proteins and DNA substrates has been accomplished, for instance, the large T antigen bound to the simian virus 40 origin of replication[75], the DNA-PKcs kinase bound to a double-stranded DNA fragment that simulates a DNA repair signal[33,76], and the clamp-loading complex for DNA replication[77].
High resolution structures Averaging of the noise images from single-particle EM has the potential to deliver 3D structures with a resolution sufficient to trace the polypeptide chain[50]. A Nature paper published in 2003 on the flagellar structure[78] demonstrated that if good single images can be accurately aligned, they are able to deliver atomic information. The authors made use of a trick to align the images according to their regular arrangement within the flagella. Nevertheless, the outstanding merit of the work was that no diffraction information was used during their processing. It seems clear that reaching very high resolution will only be possible for adequate specimens of large size, high symmetry and by means of high-quality (ie high signal-to-noise ratio) images, derived probably from helium-cooled microscopes. However, the race is on to increase the resolution of single-particle reconstructions both with improved equipment and better algorithms. Some recent examples are the GroEL structure at 6 Å[19] using EMAN, the Escherichia coli large ribosomal subunit at 7.5 Å[79], and the 8 Å resolution structure of microtubules[41] using SPIDER.
Other noteworthy works Other recent studies of special interest have been the definition of the structural basis of pore formation by the bacterial toxin pneumolysin, by the group of Helen Saibil[80], and the pseudo-atomic model of the capsid of phage phi29[34].
Future prospects and limitations of single-particle electron microscopy
Single-particle EM analysis has become a trendy structural tool in biology despite not providing atomic resolution information. This is due to some of the great advantages of this method in comparison to more established atomic resolution techniques: (i) small amounts of purified protein are required, compatible with those ordinarily obtained for large macromolecules or multi-subunit protein complexes, possibly its key advantage when compared to crystallography and NMR as EM can deliver biological information from modest quantities of material; (ii) macromolecules are trapped in their native conformation in physiological buffers; and (iii) preparations containing mixed populations or contaminated samples could be potentially analyzed whenever the distinct populations can be separated, either visually or computationally. Consequently, single-particle EM is a technique very suitable for determining the 3D structure of large macromolecule complexes (“molecular machines”), which are now known to be implicated in many cellular processes. This is so because macromolecular complexes can be very large and challenging to crystallize; they can frequently be purified only in modest quantities, while at the same time being very flexible and of variable composition.
Future developments of the technique are being directed toward improved resolution and a more profound examination of the volumetric data by either the fitting of atomic structures or the identification of folds. All of these advances will require new methods now under development[7,13,81]. Automation of data collection and analysis is also becoming an important goal for EM. The long learning process needed to properly operate a modern electron microscope, to collect good-quality data and to perform a flawless image processing means that only those with extended experience in EM can really do the job. Hence, making as many of those processes as automatic as possible is a great need for the future development of EM, especially when collecting the many thousands of single images needed when high resolutions are the goal. Some interesting approximations are already under development[82–87], and there are no serious conceptual reasons why automation of the most repetitive microscope tasks should not be achieved in the near future. However, one of the more important challenges for the future will be to deal with conformational flexibility and the heterogeneity of large protein complexes. As resolution increases, more data on identical conformations must be averaged, but the better the resolution, the more likely it is that molecules will differ in their exact conformation. Suggested solutions implicate the refinement of the data into more than one possible 3D volume and the exploration of the conformational space of a protein using normal-mode analysis[88–91].
Still, EM analysis presents some important limitations that are essential to keep in mind. To begin with, the methods we use today are still very much dependent on the expertise of the user to deliver the correct structure, especially when dealing with small and low-symmetry molecules or heterogeneous samples. Things can be done wrong, and an inexperienced user can end up in local minima not representing the real 3D structure. Consequently, a great challenge for the near future should be to standardize methods and controls as is done in modern X-ray crystallography. A further limitation to the method is that, in some cases, structures of macromolecular complexes are solved only to moderate resolutions, which might provide few or no biologically relevant information, especially when no atomic data of any part of a complex is known. Nevertheless, in these cases, EM structures can still be interpreted by calculating difference maps among several reconstructions to then determine the position of components in the complex. Besides these limitations, single-particle EM will certainly cope with its future challenges to become a widespread method to study the 3D structures of macromolecules in conjunction with other structural techniques.
Acknowledgement
I greatly acknowledge the constant support by Laurence H Pearl at the Institute of Cancer Research (London, UK) in the DNA-PK work presented in the figures of this review. I am also very thankful for the work of Angel Rivera-Calzada and Ernesto Arias-Palomo in my lab. I apologize to all those groups working in the EM field whose studies have not been reflected in this mini review, which was just intended as an initiation for those outside of our discipline.
References
- Baumeister W, Steven AC. Macromolecular electron microscopy in the era of structural genomics. Trends Biochem Sci 2000;25:624-31.
- Saibil HR. Macromolecular structure determination by cryo-electron microscopy. Acta Crystallogr D Biol Crystallogr 2000;56:1215-22.
- Saibil HR. Conformational changes studied by cryo-electron microscopy. Nat Struct Biol 2000;7:711-4.
- Orlova EV, Saibil HR. Structure determination of macromolecular assemblies by single-particle analysis of cryo-electron micrographs. Curr Opin Struct Biol 2004;14:584-90.
- Frank J. Single-particle imaging of macromolecules by cryo-electron microscopy. Annu Rev Biophys Biomol Struct 2002;31:303-19.
- Frank J, Schlichting I. Time-resolved imaging of macromolecular processes and interactions. J Struct Biol 2004;147:209-10.
- van Heel M, Gowen B, Matadeen R, Orlova EV, Finn R, Pape T, et al. Single-particle electron cryo-microscopy: towards atomic resolution. Q Rev Biophys 2000;33:307-69.
- van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy 1987;21:111-23.
- Nogales E, Grigorieff N. Molecular machines: putting the pieces together. J Cell Biol 2001;152:F1-10.
- Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification–powerful tools in modern electron microscopy. Biol Proced Online 2004;6:23-34.
- Viadiu H, Stemmann O, Kirschner MW, Walz T. Domain structure of separase and its binding to securin as determined by EM. Nat Struct Mol Biol 2005;12:552-3.
- Llorca O, Arias-Palomo E, Zugaza JL, Bustelo XR. Global conformational rearrangements during the activation of the GDP/GTP exchange factor Vav3. Embo J 2005;24:1330-40.
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 2003;142:334-47.
- Velazquez-Muriel JA, Sorzano CO, Fernandez JJ, Carazo JM. A method for estimating the CTF in electron microscopy based on ARMA models and parameter adjustment. Ultramicroscopy 2003;96:17-35.
- Penczek PA, Zhu J, Frank J. A common-lines based method for determining orientations for N>3 particle projections simultaneously. Ultramicroscopy 1996;63:205-18.
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 1999;128:82-97.
- Sorzano CO, Marabini R, Boisset N, Rietzel E, Schroder R, Herman GT, et al. The effect of overabundant projection directions on 3D reconstruction algorithms. J Struct Biol 2001;133:108-18.
- Sewell BT, Best RB, Chen S, Roseman AM, Farr GW, Horwich AL, et al. A mutant chaperonin with rearranged inter-ring electrostatic contacts and temperature-sensitive dissociation. Nat Struct Mol Biol 2004;11:1128-33.
- Ludtke SJ, Chen DH, Song JL, Chuang DT, Chiu W. Seeing GroEL at 6 A resolution by single particle electron cryomicroscopy. Structure (Camb) 2004;12:1129-36.
- Radermacher M. Three-dimensional reconstruction of single particles from random and nonrandom tilt series. J Electron Microsc Tech 1988;9:359-94.
- Golas MM, Sander B, Will CL, Luhrmann R, Stark H. Major conformational change in the complex SF3b upon integration into the spliceosomal U11/U12 di-snRNP as revealed by electron cryomicroscopy. Mol Cell 2005;17:869-83.
- Carazo JM, Wagenknecht T, Frank J. Variations of the three-dimensional structure of the Escherichia coli ribosome in the range of overlap views. An application of the methods of multicone and local single-cone three-dimensional reconstruction. Biophys J 1989;55:465-77.
- Crowther RA, Amos LA, Finch JT, De Rosier DJ, Klug A. Three dimensional reconstructions of spherical viruses by Fourier synthesis from electron micrographs. Nature 1970;226:421-5.
- Crowther RA, Klug A. Structural analysis of macromolecular assemblies by image reconstruction from electron micrographs. Annu Rev Biochem 1975;44:161-82.
- Bottcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 1997;386:88-91.
- Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997;386:91-4.
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 1996;116:190-9.
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol 1996;116:17-24.
- Conway JF, Steven AC. Methods for reconstructing density maps of “single” particles from cryoelectron micrographs to subnanometer resolution. J Struct Biol 1999;128:106-18.
- Sorzano CO, Marabini R, Velazquez-Muriel J, Bilbao-Castro JR, Scheres SH, Carazo JM, et al. XMIPP: a new generation of an open-source image processing package for electron microscopy. J Struct Biol 2004;148:194-204.
- Scheres SH, Valle M, Nunez R, Sorzano CO, Marabini R, Herman GT, et al. Maximum-likelihood multi-reference refinement for electron microscopy images. J Mol Biol 2005;348:139-49.
- Scheres SH, Marabini R, Lanzavecchia S, Cantele F, Rutten T, Fuller SD, et al. Classification of single-projection reconstructions for cryo-electron microscopy data of icosahedral viruses. J Struct Biol 2005;151:79-91.
- Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure (Camb) 2005;13:243-55.
- Morais MC, Choi KH, Koti JS, Chipman PR, Anderson DL, Rossmann MG. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of phi29. Mol Cell 2005;18:149-59.
- Wriggers W, Chacon P. Modeling tricks and fitting techniques for multiresolution structures. Structure (Camb) 2001;9:779-88.
- Chacon P, Wriggers W. Multi-resolution contour-based fitting of macromolecular structures. J Mol Biol 2002;317:375-84.
- Jiang W, Baker ML, Ludtke SJ, Chiu W. Bridging the information gap: computational tools for intermediate resolution structure interpretation. J Mol Biol 2001;308:1033-44.
- Rossmann MG, Morais MC, Leiman PG, Zhang W. Combining x-ray crystallography and electron microscopy. Structure (Camb) 2005;13:355-62.
- Chiu W, Baker ML, Jiang W, Dougherty M, Schmid MF. Electron cryomicroscopy of biological machines at subnanometer resolution. Structure (Camb) 2005;13:363-72.
- Rossmann MG, Mesyanzhinov VV, Arisaka F, Leiman PG. The bacteriophage T4 DNA injection machine. Curr Opin Struct Biol 2004;14:171-80.
- Li H, DeRosier DJ, Nicholson WV, Nogales E, Downing KH. Microtubule structure at 8 A resolution. Structure (Camb) 2002;10:1317-28.
- Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, et al. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature 2004;432:573-9.
- Wriggers W, Milligan RA, McCammon JA. Situs: a package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol 1999;125:185-95.
- Rossmann MG, Bernal R, Pletnev SV. Combining electron microscopy with X-ray crystallographic structures. J Struct Biol 2001;136:190-200.
- Hinsen K, Reuter N, Navaza J, Stokes DL, Lacapere JJ. Normal mode-based fitting of atomic structure into electron density maps: application to sarcoplasmic reticulum Ca-ATPase. Biophys J 2005;88:818-27.
- Navaza J, Lepault J, Rey FA, Alvarez-Rua C, Borge J. On the fitting of model electron densities into EM reconstructions: a reciprocal-space formulation. Acta Crystallogr D Biol Crystallogr 2002;58:1820-5.
- Roseman AM. Docking structures of domains into maps from cryo-electron microscopy using local correlation. Acta Crystallogr D Biol Crystallogr 2000;56:1332-40.
- Wriggers W, Birmanns S. Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. J Struct Biol 2001;133:193-202.
- Velazquez-Muriel JA, Sorzano CO, Scheres SH, Carazo JM. SPI-EM: towards a tool for predicting CATH superfamilies in 3D-EM maps. J Mol Biol 2005;345:759-71.
- Henderson R. The potential and limitations of neutrons, electrons and x-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys 1995;28:171-93.
- Okorokov AL, Orlova EV, Kingsbury SR, Bagneris C, Gohlke U, Williams GH, et al. Molecular structure of human geminin. Nat Struct Mol Biol 2004;11:1021-2.
- Rodal AA, Sokolova O, Robins DB, Daugherty KM, Hippenmeyer S, Riezman H, et al. Conformational changes in the Arp2/3 complex leading to actin nucleation. Nat Struct Mol Biol 2005;12:26-31.
- Rodal AA, Sokolova O, Robins DB, Daugherty KM, Hippenmeyer S, Riezman H, et al. Structure and molecular organization of mammalian fatty acid synthase. Nat Struct Mol Biol 2005;12:225-32.
- Jiang QX, Wang DN, MacKinnon R. Electron microscopic analysis of KvAP voltage-dependent K+ channels in an open conformation. Nature 2004;430:806-10.
- Skiniotis G, Boulanger MJ, Garcia KC, Walz T. Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor. Nat Struct Mol Biol 2005;12:545-51.
- Nakagawa T, Cheng Y, Ramm E, Sheng M, Walz T. Structure and different conformational states of native AMPA receptor complexes. Nature 2005;433:545-9.
- Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J. Locking and unlocking of ribosomal motions. Cell 2003;114:123-34.
- Valle M, Gillet R, Kaur S, Henne A, Ramakrishnan V, Frank J. Visualizing tmRNA entry into a stalled ribosome. Science 2003;300:127-30.
- Frank J. Electron microscopy of functional ribosome complexes. Biopolymers 2003;68:223-33.
- Rawat UB, Zavialov AV, Sengupta J, Valle M, Grassucci RA, Linde J, et al. A cryo-electron microscopic study of ribosome-bound termination factor RF2. Nature 2003;421:87-90.
- Halic M, Becker T, Pool MR, Spahn CM, Grassucci RA, Frank J, et al. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 2004;427:808-14.
- Klaholz BP, Myasnikov AG, van Heel M. Visualization of release factor 3 on the ribosome during termination of protein synthesis. Nature 2004;427:862-5.
- Klaholz BP, Pape T, Zavialov AV, Myasnikov AG, Orlova EV, Vestergaard B, et al. Structure of the Escherichia coli ribosomal termination complex with release factor 2. Nature 2003;421:90-4.
- Stark H, Rodnina MV, Wieden HJ, Zemlin F, Wintermeyer W, van Heel M. Ribosome interactions of aminoacyl-tRNA and elongation factor Tu in the codon-recognition complex. Nat Struct Biol 2002;9:849-54.
- Jurica MS, Sousa D, Moore MJ, Grigorieff N. Three-dimensional structure of C complex spliceosomes by electron microscopy. Nat Struct Mol Biol 2004;11:265-9.
- Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. RNA 2002;8:426-39.
- Boehringer D, Makarov EM, Sander B, Makarova OV, Kastner B, Luhrmann R, et al. Three-dimensional structure of a pre-catalytic human spliceosomal complex B. Nat Struct Mol Biol 2004;11:463-8.
- Golas MM, Sander B, Will CL, Luhrmann R, Stark H. Molecular architecture of the multiprotein splicing factor SF3b. Science 2003;300:980-4.
- Wu PY, Ruhlmann C, Winston F, Schultz P. Molecular architecture of the S cerevisiae SAGA complex. Mol Cell 2004;5:199-208.
- Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 2002;9:423-32.
- Llorca O, McCormack EA, Hynes G, Grantham J, Cordell J, Carrascosa JL, et al. Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature 1999;402:693-6.
- Llorca O, Martin-Benito J, Ritco-Vonsovici M, Grantham J, Hynes GM, Willison KR, et al. Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J 2000;19:5971-9.
- Martin-Benito J, Boskovic J, Gomez-Puertas P, Carrascosa JL, Simons CT, Lewis SA, et al. Structure of eukaryotic prefoldin and of its complexes with unfolded actin and the cytosolic chaperonin CCT. EMBO J 2002;21:6377-86.
- Martin-Benito J, Boskovic J, Gomez-Puertas P, Carrascosa JL, Simons CT, Lewis SA, . Structure of the complex between the cytosolic chaperonin CCT and phosducin-like protein. Proc Natl Acad Sci USA 2004; 101: 17 410–5.
- Gomez-Lorenzo MG, Valle M, Frank J, Gruss C, Sorzano CO, Chen XS, et al. Large T antigen on the simian virus 40 origin of replication: a 3D snapshot prior to DNA replication. EMBO J 2003;22:6205-13.
- Boskovic J, Rivera-Calzada A, Maman JD, Chacon P, Willison KR, Pearl LH, et al. Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO J 2003;22:5875-82.
- Miyata T, Oyama T, Mayanagi K, Ishino S, Ishino Y, Morikawa K. The clamp-loading complex for processive DNA replication. Nat Struct Mol Biol 2004;11:632-6.
- Yonekura K, Maki-Yonekura S, Namba K. Complete atomic model of the bacterial flagellar filament by electron cryomicroscopy. Nature 2003;424:643-50.
- Matadeen R, Patwardhan A, Gowen B, Orlova EV, Pape T, Cuff M, et al. The Escherichia coli large ribosomal subunit at 7.5 A resolution. Structure Fold Des 1999;7:1575-83.
- Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 2005;121:247-56.
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 2003;333:721-45.
- Rath BK, Frank J. Fast automatic particle picking from cryo-electron micrographs using a locally normalized cross-correlation function: a case study. J Struct Biol 2004;145:84-90.
- Plaisier JR, Koning RI, Koerten HK, van Heel M, Abrahams JP. TYSON: robust searching, sorting, and selecting of single particles in electron micrographs. J Struct Biol 2004;145:76-83.
- Booth CR, Jiang W, Baker ML, Zhou ZH, Ludtke SJ, Chiu W. A. 9 angstroms single particle reconstruction from CCD captured images on a 200 kV electron cryomicroscope. J Struct Biol 2004;147:116-27.
- Nickell S, Forster F, Linaroudis A, Net WD, Beck F, Hegerl R, et al. TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol 2005;149:227-34.
- Lei J, Frank J. Automated acquisition of cryo-electron micrographs for single particle reconstruction on an FEI Tecnai electron microscope. J Struct Biol 2005;150:69-80.
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, et al. Automated molecular microscopy: the new Leginon system. J Struct Biol 2005;151:41-60.
- Tama F, Wriggers W, Brooks CL. Exploring global distortions of biological macromolecules and assemblies from low-resolution structural information and elastic network theory. J Mol Biol 2002;321:297-305.
- Chacon P, Tama F, Wriggers W. Mega-Dalton biomolecular motion captured from electron microscopy reconstructions. J Mol Biol 2003;326:485-92.
- Wriggers W, Chakravarty S, Jennings PA. Control of protein functional dynamics by peptide linkers. Biopolymers 2005. in press.
- Carazo JM. Accessing information on the conformational flexibility of molecular machines. Structure (Camb) 2004;12:170-1.