
Effects of simvastatin on cardiac performance and expression of sarcoplasmic reticular calcium regulatory proteins in rat heart1
Introduction
There is evidence that early initiation of statins, HMG-CoA reductase inhibitors, in patients is associated with markedly reduced fatal or non-fatal myocardial infarction and the mortality of cardiovascular event. Scandinavian Simvastatin Survival Study (4S) and Heart Protect Study (HPS) test have demonstrated that simvastatin could decrease the incidence of ischemic stroke, myocardial infarction, and the mortality of severe cardiovascular events in atherosclerotic and hypercholesterolemic patients[1,2]. However, it is unclear whether treatment with simvastatin would influence cardiac function.
It is well known that Ca2+ plays a central role in excitation-contraction coupling of myocardium. Intracellular Ca2+ ([Ca2+]i) is from both of release of Ca2+ from intracellular Ca2+ stores and influx of extracellular Ca2+ across the plasma membrane. Several main proteins in sarcoplasmic reticulum (SR), such as SERCA and RyR2, are involved in the release, uptake, and storage of cardiac muscle Ca2+. Contraction is mediated through the release of Ca2+ from the SR by IP3R (Ca2+ pool sensitive to IP3) and RyR (Ca2+ pool insensitive to IP3), while relaxation involves the active re-uptake of Ca2+ into the SR lumen by sarcoplasmic reticulum Ca2+-ATPase (SERCA). In cardiac muscle, the SERCA activity is under reversible regulation by phospholamban (PLB).
As previous studies revealed, simvastatin induced an increase in [Ca2+]i through two different pathways (ie the Ca2+ release from intracellular stores sensitive to thapsigargin and ryanodine and the Ca2+ influx from extracellular solution in endothelial cells[3,4], L6 rat myoblasts[5], or cultured cardio-myocytes[6]). In some pathophysiological conditions, statins can prevent Ca2+ overload. For example, liposoluble HMG-CoA reductase inhibitors could markedly inhibit [Ca2+]i induced by angiotensin II, LPC, and noradrenaline[7–9]. Atrovastatin prevented the enhanced uptake of Ca2+ by sarcoplasmic reticulum (SR) and non-SR Ca2+ stores in diabetic dyslipi-demic pigs[10]. However, it is still unclear whether simvastatin can induce the alteration of expression of SERCA, PLB, and RyR2. Thus, the present study was designed to evaluate the effect of simvastatin on cardiac contractile function and gene and protein expression of SR calcium regulatory proteins, such as SERCA, PLB, and RyR2, which are related with the alteration of [Ca2+]i.
Materials and methods
Drugs Simvastatin (gifted by Merck Sharp & Dohme, Hertfordshire, UK), a lactone prodrug, was diluted in 0.5 mL of 100% ethanol, mixed with 0.75 mL of 0.1 mol/L NaOH, heated at 50 °C for 2 h, neutralized with 0.1 mol/L HCl to pH 7.2, adjusted with deionized water to a final concentration of 5 mmol/L, sterilized by filtration, and stored in aliquots at -20 °C[11]. Other main reagents in this experiment include: Trizol (Invitrogen, CA, USA); MMLV and pUC19 DNA/MspI (HpaII) Marker (MBI Fermentas, Vilnius, Lithuania); Taq plus DNA polymerase and primers (Sangon, Shanghai, China); DMEM (Gibco, Invitrogen); trypsin 1:250 (Amresco, Solon OH, USA); collagenase I (Sigma, St Louis, MO, USA); 5-bromo-2-deoxyuridine (Sigma); fetal bovine serum (Hang-zhou-Sijiqing Biological Engineering Materials Co Ltd, China); rabbit anti-rat myosin heavy chain antibody (Novocastia Lab Ltd, USA); FITC-conjugated goat anti-rabbit IgG (Amersham, Buckinghamshire, UK); monoclonal mouse antibody to SERCA, PLB and RyR2 (ABR corporation); secondary anti-goat antibody (Beijing Zhongshan Biotechnology, Beijing, China); MTT (Sigma); EZ-ECL Chemiluminescence Detection Kit for HRP (Biological Industries, Haemek, Israel).
Animals Male Sprague-Dawley (SD) rats (240–270 g) and 1–3 d old SD rats were purchased from the Experimental Animal Center, Chinese Academy of Sciences (Shanghai, China) and all procedures in our experiments were approved by the Animal Care and Use Committee of Zhejiang Univer-sity.
Isolated heart perfusion Rats were anesthetized with sodium pentobarbital (60 mg/kg), then the hearts were excised and perfused in a non-recirculating Langendorff apparatus with modified Krebs-Henseleit (KH) solution (in mmol/L: NaCl 118, KCl 4.7, CaCl2 1.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25.2, and glucose 11.1, pH 7.4). The coronary perfusion pressure was 100 mmHg and KH solution was bubbled with 95% O2:5% CO2 at 37 °C. A water-filled latex balloon-tipped catheter was inserted into the left ventricle via the left atrium and connected to a computer coupled with MedLab software (Nanjing MedEase Science & Technology Co Ltd, China) through a pressure transducer. The volume of the balloon was adjusted to a left ventricular end-diastolic pressure of 6–8 mmHg during the initial equilibration, then remained constant throughout the experiment. Left ventricular developed pressure (LVDP) and the maximum rate of intraventricular pressure development and relaxation (±dp/dtmax) were measured during perfusion.
All hearts were equilibrated for 15 min for baseline measurements, and were then subjected to 60 min of reperfu-sion with or without simvastatin at 1, 3, 10, 30, or 100 µmol/L. After perfusion, the hearts were removed with the right ventricle and connective tissues, freeze-clamped into liquid nitrogen, then stored at -80 °C.
Cell culture Primary cultures of neonatal rat ventricular cardiomyocytes from 1–3 d old SD rats were performed as described previously[12]. Briefly, the ventricle was dissociated with the admixture of 0.25% trypsin and 0.1% collagenase I, then cell suspensions were washed with DMEM supplemented with 20% fetal bovine serum (FBS) and centrifuged at 1000 rpm for 6 min. The centrifuged cells were then resuspended in 10% FBS containing DMEM. These cells were preplated for 1 h in order that nonmyocyte readily attached to the bottom of the culture dishes. Unattached cells, which were enriched with cardiac myocytes, were plated onto 6-well dishes at a density of 0.5×106 cells/well. BrdU (0.01 mmol/L) was added during the first 72 h to prevent proliferation of nonmyocytes[12] (Figure 1).
Myocardial cells were incubated in DMEM containing non-bovine serum albumin for 24 h prior to respective simvastatin (1, 3, 10, 30, or 100 µmol/L) treatment for 1 h or 24 h.
RT-PCR Total RNA was extracted using TRIzol reagent from rat heart tissues and cultured neonatal rat ventricular cardiomyocytes according to the manufacturer’s instruction. First-strand cDNA was generated by adding 2 µg total RNA, 5 µL of 5×reverse transcriptase buffer, 3 µL (0.5 µg/µL) randomhexamer primers, 2 µL (10 mmol/L) dNTP, 1 µL (200 U/μL) M-MLV reverse transcriptase and autoclaved water in a final volume of 20 µL. Reverse transcription was carried out at 42 °C for 90 min, followed by heat inactivation at 70 °C for 5 min, terminated at 70 °C for 10 min. Then, PCR was run in a DNA thermal cycler (PTC150 Thermo-Cycler, MJ Research, Inc, Waltham, MA, USA). The primers were up primer 5'-AAG-CAG-TTC-ATC-CGC-TAC-CT-3' and down primer 5'-AGA-CCA-TCC-GTC-ACC-AGA-TT-3' for SERCA; up primer 5'-TAC-CTT-ACT-CGC-TCG-GCT-ATC -3' and down primer 5'-CAG-AAG-CAT-CAC-AAT-GAT-GCA-G-3' for PLB; and up primer 5'-ACT-GCT -AAA-GTG-ACC-AAC-AG-3' and down primer 5'-TTG-CAT-CGC-TGA-AAT-CTA-GT-3' for RyR2. The β-actin gene (up primer 5'-GAG-ACC-TTC-AAC-ACC-CCA-GCC-3', down primer 5'-GGC-CAT-CTC-TTG-CTC-GAA-GTC-3') was used as control. Taq polymerase 1.5 units, 2 µL cDNA, 1 µL (10 µmol/L) each primer, 1 µL (10 µmol/L) β-actin, 1 µL of 10 mmol/L dNTP solution, 2.5 µL PCR buffer solution and ultrapure autoclaved water were used to reach a total reaction volume of 25 µL. The cycling parameters were as follows: 5 min of 1 cycle at 94 °C, 45 s at 94 °C, 1 min at 57°C, and 1 min of 23 cycles at 72 °C (SERCA); 30 sec at 94 °C, 1 min at 55 °C, and 1 min of 24 cycles at 72 °C (PLB); 45 s at 94 °C, 1 min at 57 °C, and 1 min of 27 cycles at 72 °C (RyR2); and 10 min of 1 cycle at 72 °C. The gray scales of absorbance in goal gene and β-actin gene were determined and assayed with Kodak digital science Electrophoresis Documentation and Analysis System120.
Western blot Heart tissue 50 mg was homogenized in 1 mL modified tonic sucrose[13] (0.3 mol/L sucrose, 10 mmol/L imidazole,10 mmol/L sodium metabisulfite, 1 mmol/L DTT, 0.3 mmol/L PMSF) and centrifuged at 13000×g at 4 °C for 15 min. Cultured cells were scraped in ice-cold RIPA solubilization buffer consisting of 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaF, 1% sodium deoxycholate, 1% Nodinet-P40, 0.1% sodium dodecylsulfate, 2.5 mmol/L pretreated sodium orthovanadate, 125 µmol/L phenylarsine oxide, and 2 mmol/L phenylmethyl sulphonyl fluoride[14] and centrifuged at 14 000×g for 30 min. The protein concentration was determined by the Brandford method.
Aliquots containing 24 µg of protein for SERCA or 10 µg of protein for PLB were loaded on the SDS-polyacrylamide gel and separated by electrophoresis (6% or 10% acrylamide separating gel, respectively, for 30 min). The separated proteins were electrophoretically transferred onto nitrocellulose membranes (Amersham) at 200 mA at 4 °C for 90 min in a buffer containing 25 mmol/L Tris base, 192 mmol/L glycine, and 20% methanol by using the Bio-Rad Trans-Blot electrophoretic transfer system (Bio-Rad). Aliquots of 6 μg RyR2 SDS-digested homogenate protein were loaded on the SDS-polyacrylamide gel, separated by electrophoresis (4% acrylamide stacking gel and 6% acrylamide separating gel), and were electrophoretically transferred onto polyvinylidene difluoride membranes (Bio-Rad) at 250 mA at 4 °C for 3 h and then at 50 V for about 12 h. These membranes were incubated at 4 °C overnight with mouse-anti-SERCA, mouse-anti-PLB, or mouse-anti-ryanodine receptor at 1:1000 dilution in 5% Carnation instant milk/TBS. After incubation with a secondary anti-goat antibody at a 1:500 dilution in 5% Carnation instant milk-TBS-Tween 20, the blot was developed using enhanced chemiluminescence according to the manual (Beit Haemek LTD) and exposed to X-ray film[11].
MTT assay Cells were incubated in the absence or presence of simvastatin for 24 h, and then the cytotoxic effect was assessed using MTT assay method. Each well of plates was added with 10 µL MTT (5 g /L) per 100 µL of medium, and were incubated at 37 °C for 4 h. Then, the medium in each well was replaced with 100 µL dimethylsulfoxide and the plates were incubated for 5–10 min at room temperature for color development. Finally, 96-well of plates was read by enzyme-linked immunosorbent assay (ELISA) reader (570 nm) to get the absorbance density values[15].
Statistics Data were presented as mean±SD. To correct for variability in basal values, changes in left ventricular development pression, +dp/dtmax and -dp/dtmax were calculated as a percent of the initial basal values for each heart. Various kinds of indexes between the control group and simvastatin-treated groups were analyzed by one way ANOVA with SPSS 10.0 software. P<0.05 was considered the threshold for statistical significance between the control group and the experimental groups.
Results
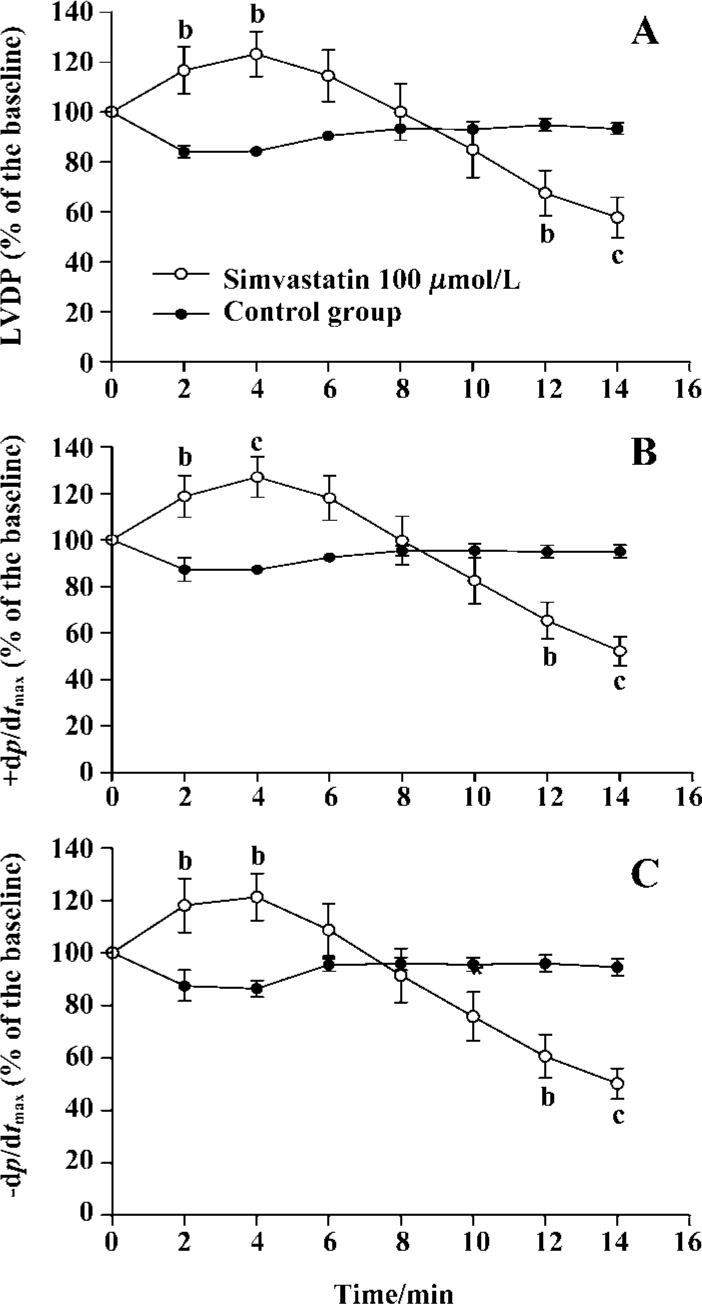
Effect of simvastatin on cardiac performance To determine whether simvastatin exerted any direct cardiodynamic effect, the isolated rat hearts were initially perfused without or with simvastatin for 60 min in this experiment. The results showed that simvastatin 1 µmol/L had no impact on cardiac performance in the isolated rat hearts compared with control (LVDP: 68.9%±17.0% vs 71.7%±10.6%, P>0.05; +dp/dtmax: 69.3%±5.3% vs 72.1%±7.5%, P>0.05; -dp/dtmax: 67.3%±5.5% vs 68.5%±6.1%, P>0.05). However, simvastatin 3, 10, and 30 µmol/L significantly increased the levels of LVDP and ±dp/dtmax of rat hearts compared with those of control (LVDP: 95.8%±17.4%, 85.9%±8.6%, 96.9%±7.0% vs 71.7%±10.6%, P<0.01, 0.05, 0.01; +dp/dtmax: 89.5%±4.4%, 97.2%±10.8%, 89.8%±6.7% vs 72.1%±7.5%, P<0.01 all; -dp/dtmax: 82.9%±4.7%, 88.6%±11.0%, 79.6%±6.6% vs 68.5%±6.1%, P<0.01 all, respectively).
When 100 µmol/L simvastatin was applied in isolated rat hearts, sudden arrest of the isolated heart, following transient increased contractile performance, occurred. The scope of sudden death time interval was from 14 to 20 min. Thus, the measurement of LVDP% and ±dp/dtmax% were recorded every 2 min until 14 min (Figure 2).
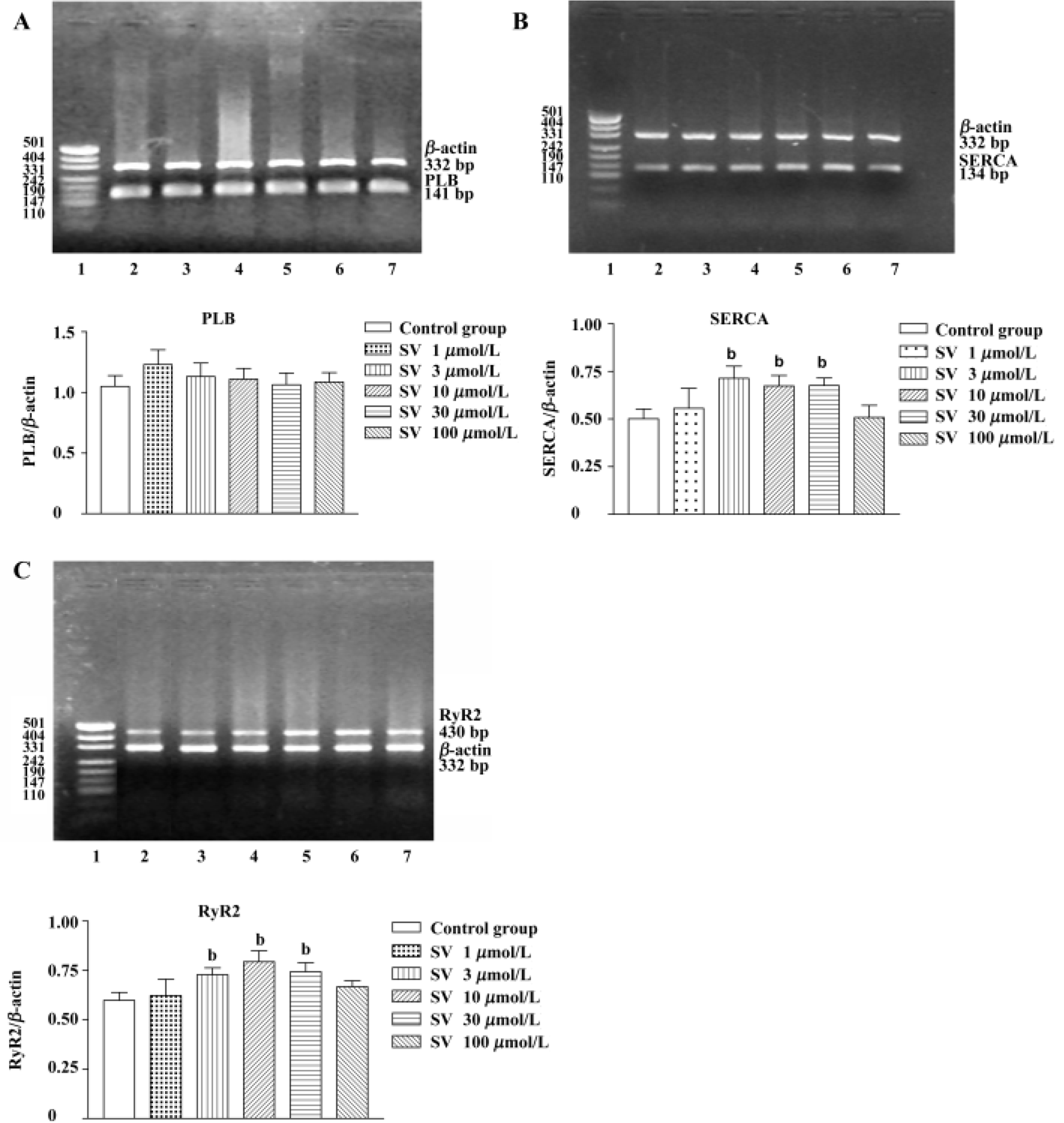
Gene expression of SR protein in isolated rat hearts and cultured cardiomyocytes After being treated with simvastatin at different concentrations (3, 10, and 30 µmol/L), SERCA and RyR2 mRNA levels of isolated heart tissue were increased. However, no alteration was found in heart tissue after simvastatin 1 or 100 µmol/L treatment. There was no difference in PLB mRNA levels of rat hearts between the control group and simvastatin-treatment groups (Figure 3).
Simvastatin 3−100 µmol/L had no influence on PLB gene expression of neonatal rat ventricular cardiomyocytes, but markedly increased SERCA and RyR2 gene expression (Figure 4).
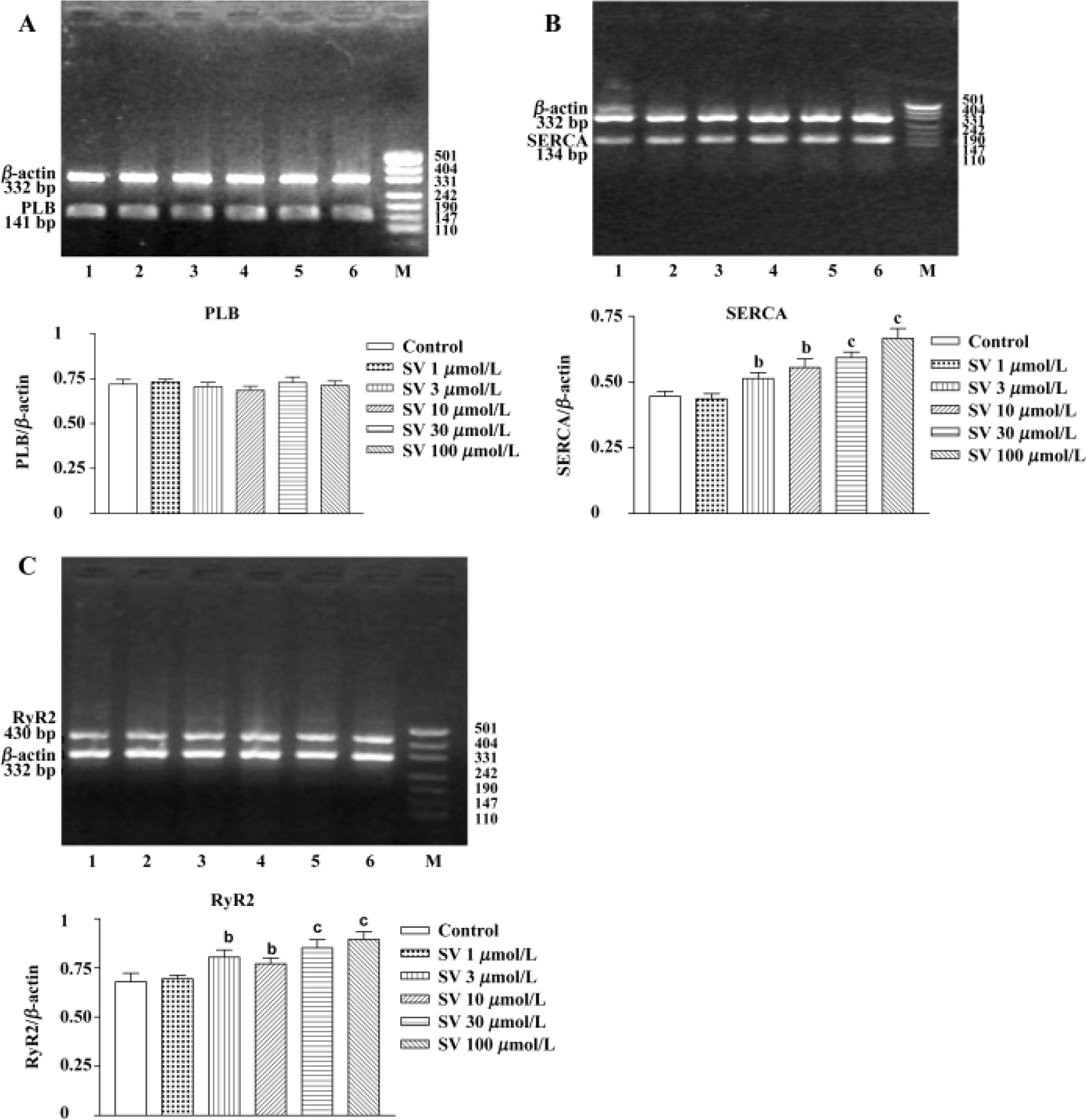
Additionally, sequencing analysis showed 100% coincidence rates of SERCA, PLB, and RyR2 cDNA sequences, which came from the final products of RT-PCR compared with those in GeneBank, indicating that the amplified fractions had high specificity.
Expression of SR protein in isolated rat hearts and cultured cardiomyocytes No alteration in the protein levels of SERCA, PLB, and RyR2 was found after simvastatin 1, 3, 10, 30, or 100 µmol/L treatment (Figure 5).
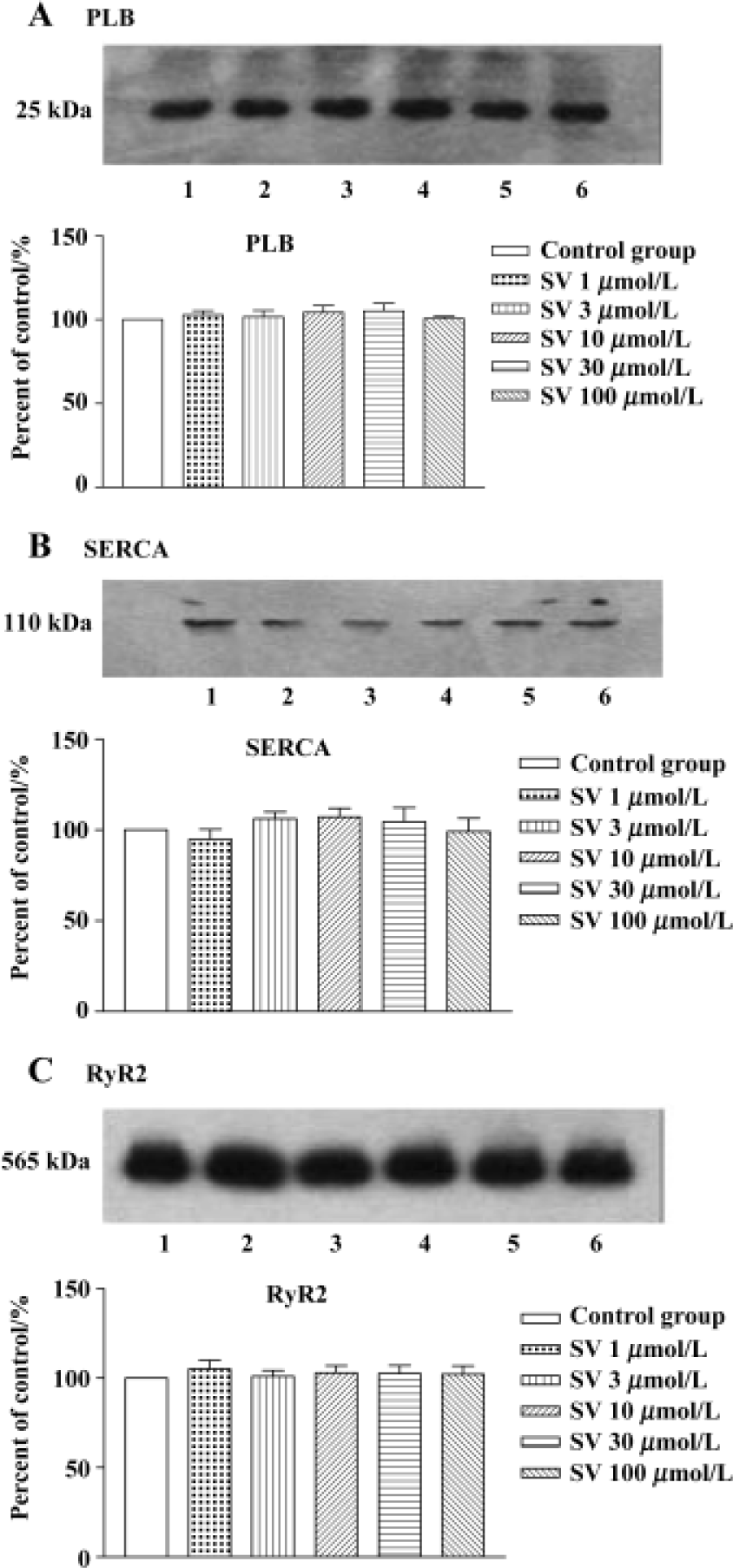
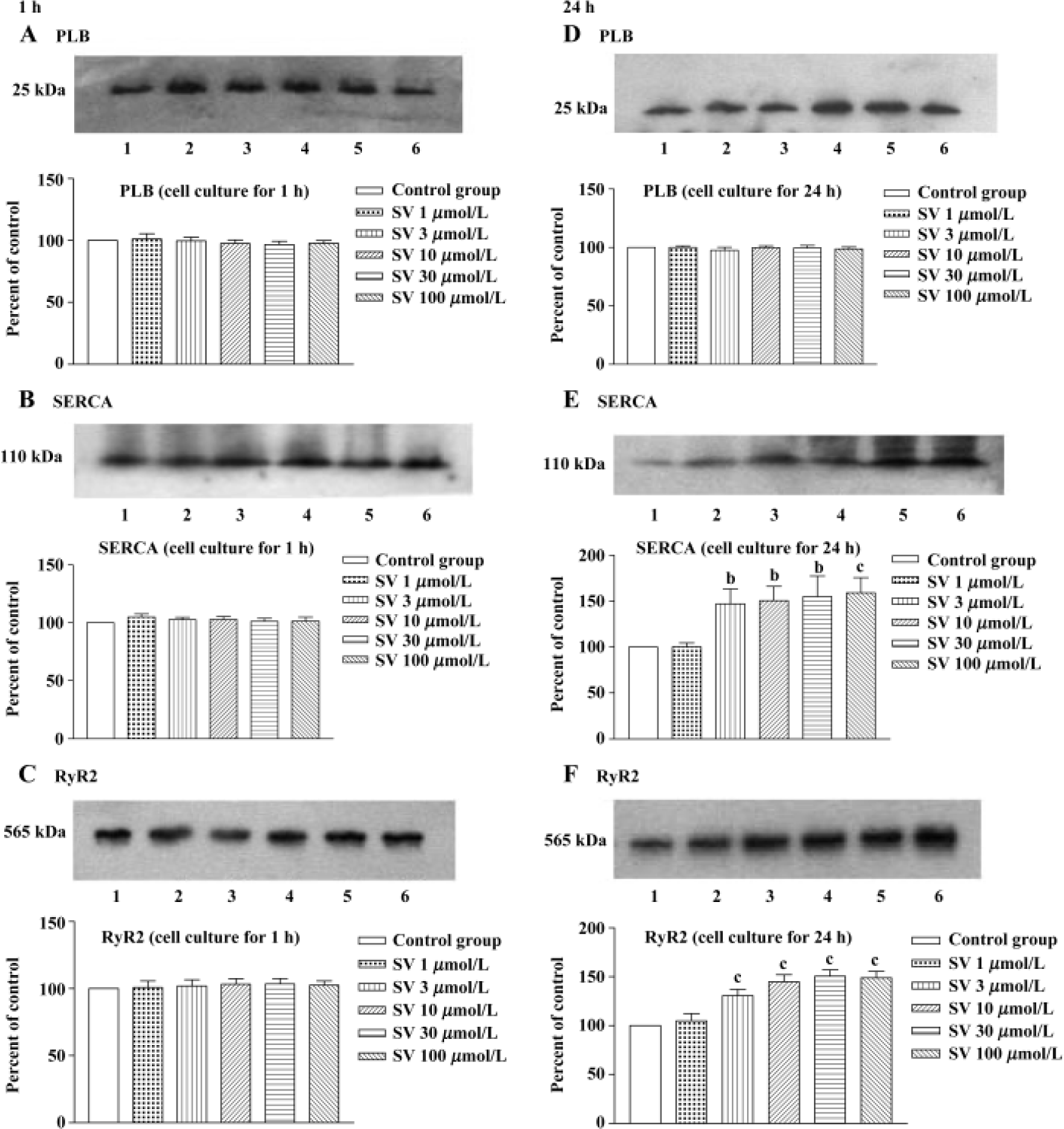
In order to test whether the changes in protein levels occurred with the elongation of simvastatin treatment, we supplied cultured cardiomyocytes with different concentration simvastatin for 1 h and 24 h. The results showed that incubation with simvastatin for 1 h in the cultured cardiomyocytes had no effect on the protein expression of SR calcium regulatory protein, whereas longer period (24 h) of simvastatin 3−100 μmol/L treatment increased SERCA and RyR2 protein level, except PLB protein levels of cardiomyo-cytes (Figure 6).
Effect of simvastatin on the cytotoxicity in cultured neonatal rat ventricular cardiomyocytes Simvastatin 1–30 µmol/L did not induce alteration of mitochondrial activity, whereas simvastatin 100 µmol/L produced a rapid drop of mitochondrial activity (Table 1).
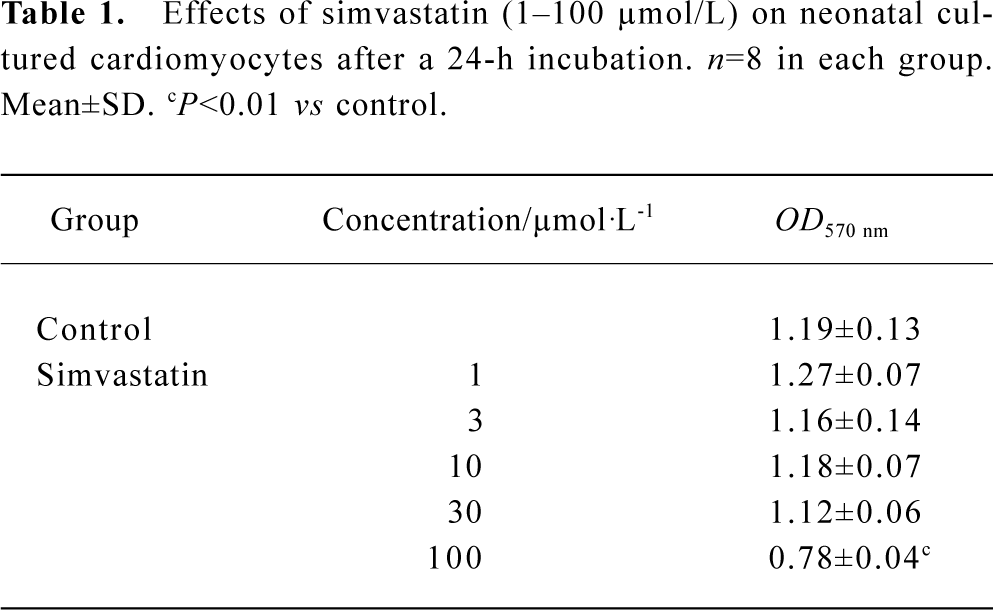
Full table
Discussion
It is well known that simvastatin can produce some beneficial effects independent of the lowering cholesterol level[16–19], so the direct pharmacological effect of simvastatin on the initial management and prevention of the disease are paid more attention. Our study was performed to identify whether simvastatin induced intrinsic alteration of cardiomyocyte contraction and related calcium regulatory protein, which might explain the effect of simvastatin on cardiac performance.
The present study demonstrated that simvastatin treatment (3, 10, and 30 µmol/L) resulted in the increase of LVDP, +dp/dtmax, and -dp/dtmax in isolated perfused hearts, while simvastatin 100 µmol/L induced sudden heart related death. It has been shown that an increase of nitrogen monoxide (NO) and a decrease of ubiquinone and oxygen-derived free radicals were involved in the possible protective mechanism in isolated rat hearts[20–22], while impairment of mitochondria was associated with injury mechanism in ischemic rat hearts[23]. Previous studies also reported[6] that under normoxic conditions simvastatin caused an increase in [Ca2+]i, which is the key event in excitation-contraction coupling in cardiac myocytes. Therefore, the improvement of cardiac performance in isolated rat hearts during 1 h of perfusion can be explained by NO elevation, decrease of oxygen-derived free radicals, and an increase of [Ca2+]i and so on.
Using RT-PCR assay, we found the elevation of RyR2 and SERCA mRNA in isolated hearts treated with simvastatin for 1 h and not in those treated with vehicle. Similar to these results, in neonatal cultured cardiac myocytes, the gene levels of RyR2 and SERCA were both elevated after a 1-h incubation. As intracellular second messenger of the organism information transmission, Ca2+ is involved in some gene expression. Even transient calcium transport elevation can induce mRNA transcription for a long time. Therefore, the results in our experiment suggested that simvastatin-induced SERCA and RyR2 expression were involved in the mechanisms mentioned above. However, a 1-h treatment with simvastatin (1, 3, 10, 30, or 100 µmol/L) in the isolated hearts and cultured cardiomyocytes did not influence the protein levels of SERCA, PLB, or RyR2, while a 24-h incubation of simvastatin in cultured cardiomyocytes increased SERCA and RyR2 protein level, except PLB protein. It has been shown that several main proteins in SR, such as SERCA and RyR2, are implicated in the release, uptake, and storage of calcium in cardiac muscle. Contraction is mediated through the release of Ca2+ from the SR by ryanodine receptor, while relaxation involves the active re-uptake of Ca2+ into the SR lumen by SERCA. The SERCA activity is under reversible regulation by PLB[24]. Thereby, it is possible that although the expression of RyR2, SERCA, and PLB were not directly correlated with the cardiodynamic increase of isolated perfused hearts, simvastatin-induced SERCA and RyR2 expression may influence cardiac performance during extended periods of perfusion. Thus, there is reason to believe that alteration of gene and protein expression of SERCA and RyR2 might be a prophetic index of cardioprotective effect of simvastatin. Furthermore, the effect of simvastatin on SR calcium regulation protein gave a clue that the molecular biological mechanism could be a new approach to explain the alteration of cardiac performance affected by simvastatin in future.
In the meantime, the present study discovered that elevation of SERCA and RyR2 mRNA were shown in cultured cardiomyocytes, but not found in isolated rat hearts in the presence of simvastatin 100 µmol/L. We suspect that the result was due to insufficient expression time due to the cardiac sudden death in isolated rat hearts. The MTT assay, as an index of cell viability and cell growth[25,26], revealed that the survival rate of cardiac cells was markedly reduced with simvastatin 100 µmol/L treatment, while cell viability was not influenced by simvastatin 1, 3, 10, 30 µmol/L. Perhaps the toxic effect of overdose simvastatin promoted decrease of cardiac cells, which may indirectly explain the sudden arrest of isolated hearts with simvastatin 100 µmol/L.
Based on our research, it is concluded that simvastatin, at reasonable pharmacological concentration, would enhance cardiac performance concomitant with gene expression increase of calcium regulatory protein. Meanwhile, it should be noted that overdose of simvastatin (100 µmol/L) can produce a deterioration effect on hearts.
References
- The Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994;344:1383-9.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002;360:7-22.
- Escobales N, Castro M, Altieri PI, Sanabria P. Simvastatin releases Ca2+ from a thapsigargin-sensitive pool and inhibits InsP3-dependent Ca2+ mobilization in vascular smooth muscle cells. J Cardiovasc Pharmacol 1996;27:383-91.
- Alvarez de Sotomayor M, Andriantsitohaina R. Simvastatin and Ca2+ signaling in endothelial cells: involvement of Rho protein. Biochem Biophys Res Commun 2001;280:486-90.
- Nakahara K, Yada T, Kuriyama M, Osame M. Cytosolic Ca2+ increase and cell damage in L6 rat myoblasts by HMG-CoA reductase inhibitors. Biochem Biophys Res Commun 1994;202:1579-8.
- Bastiaanse EM, Atsma DE, Kuijpers MM, Van der Laarse A. Simvastatin-sodium delays cell death of anoxic cardiomyocytes by inhibition of the Na+/Ca2+ exchanger. FEBS Lett 1994;343:151-4.
- Tesfamariam B, Frohlich BH, Gregg RE. Differential effects of pravastatin, simvastatin, and atorvastatin on Ca2+ release and vascular reactivity. J Cardiovasc Pharmacol 1999;34:95-101.
- Alvarez de Sotomayor M, Perez-Guerrero C, Herrera MD, Marhuenda E. Effect of simvastatin on vascular smooth muscle responsiveness: involvement of Ca2+ homeostasis. Eur J Pharmacol 2001;415:217-24.
- Yokoyama K, Ishibashi T, Ohkawara H, Kimura J, Matsuoka I, Sakamoto T. HMG-CoA reductase inhibitors suppress intracellular calcium mobilization and membrane current induced by lysophosphatidylcholine in endothelial cells. Circulation 2002;105:962-7.
- Hill BJ, Dixon JL, Sturek M. Effect of atorvastatin on intracellular calcium uptake in coronary smooth muscle cells from diabetic pigs fed an atherogenic diet . Atherosclerosis 2001;159:117-24.
- Llorente-Cortes V, Martinez-Gonzalez J, Badimon L. Differential cholesteryl ester accumulation in two human vascular smooth muscle cell subpopulations exposed to aggregated LDL: effect of PDGF-stimulation and HMG-CoA reductase inhibition. Atherosclerosis 1999;144:335-42.
- Kurosaki K, Ikeda U, Maeda Y, Shimpo M, Ueno S, Shimada K. Effects of vesnarinone on nitric oxide synthesis in rat cardiac myocytes. Cardiovasc Res 1998;38:192-7.
- Zhong Y, Ahmed S, Grupp IL, Matlib MA. Altered SR protein expression associated with contractile dysfunction in diabetic rat hearts. Am J Physiol Heart Circ Physiol 2001;281:H1137-47.
- Kwak BR, van Kempen MJ, Theveniau-Ruissy M, Gros DB, Jongsma HJ. Connexin expression in cultured neonatal rat myocytes reflects the pattern of the intact ventricle. Cardiovasc Res 1999;44:370-80.
- Huang YT, Chueh SC, Teng CM, Guh JH. Investigation of ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem Pharmacol 2004;67:727-33.
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG) CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA 1998;95:8880-5.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999;100:178-84.
- Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol 2000;20:61-9.
- Bourcier T, Libby P. HMG-CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol 2000;20:556-62.
- Di Napoli P, Antonio Taccardi A, Grilli A, Spina R, Felaco M, Barsotti A. Simvastatin reduces reperfusion injury by modulating nitric oxide synthase expression: an ex vivo study in isolated working rat hearts. Cardiovasc Res 2001;51:283-93.
- Jones SP, Trocha SD, Lefer DJ. Pretreatment with simvastatin attenuates myocardial dysfunction after ischemia and chronic reperfusion. Arterioscler Thromb Vasc Biol 2001;21:2059-64.
- Wassmann S, Laufs U, Baumer AT, Muller K, Ahlbory K, Linz W. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 2001;37:1450-7.
- Satoh K, Ichihara K. Effects of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on mitochondrial respiration in ischemic rat hearts. Eur J Pharmacol 1995;292:271-5.
- Arai M, Otsu K, MacLennan DH, Alpert NR, Periasamy M. Effect of thyroid hormone on the expression of mRNA encoding sarcoplasmic reticulum proteins. Circ Res 1991;69:266-76.
- Kong JY, Rabkin SW. Mitochondrial effects with ceramide-induced cardiac apoptosis are different from those of palmitate. Arch Biochem Biophys 2003;412:196-206.
- Balligand JL, Ungureanu-Longrois D, Simmons WW, Pimental D, Malinski TA, Kapturczak M. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J Biol Chem 1994;269:27580-8.