
Gene expression profile of amyloid beta protein-injected mouse model for Alzheimer disease
Introduction
Alzheimer disease (AD) is the most common cause of dementia in the elderly, affecting 7%–10% of individuals over 65 years of age and approximately 40% of persons over 80 years old[1]. The disease is characterized behaviorally by global cognitive decline, and defined histologically by two distinguishing pathologies: amyloid plaques, which are extracellular deposits consisting mainly of aggregated amyloid beta-protein (Aβ), and neurofibrillary tangles (NFT), which are intracellular deposits consisting predominantly of hyperphosphorylated tau protein[2]. Epidemiological and molecular studies suggest that AD has multiple etiologies, including genetic mutations, susceptibility genes and environmental factors that promote formation and accumulation of insoluble Aβ and hyperphosphorylated tau. An animal model that mimics the progression of AD was developed using icv injection of Aβ into mice[3–5]. This was accompanied by impairment in learning and memory in addition to biochemical changes and neuronal degeneration.
Recently, a high-throughput technique was developed using cDNA microarrays, in which labeled RNA hybridizes to DNA molecules attached at specific locations on the surface of micrarray membrane[6]. One of the most attractive applications of microarrays is in the study of differential gene expression in disease and animal models. Microarrays are extensively employed to study global changes in gene expression in post-mortem tissues, cultured cells, animal models, and in response to drug treatment. The expression pattern of a gene provides indirect information about function, drug target and cause of a disease. In this study we used cDNA microarrays (Clontech Laboratories, Palo Alto, CA, USA) to investigate gene expression patterns in an icv Aβ mouse model for AD in the cerebral cortex. This approach provided an additional insight into gene processes, which occur not only in AD animal model but also in other neurodegenerative disorders. The knowledge of specific cascades of events leading and causing neurodegeneration will be the key factor in developing and using neuroprotective drugs.
Materials and methods
Reagents Aβ25-35 (Sigma Chemical, St Louis, MO, USA) was dissolved in sterile normal saline at a concentration of 1 g/L and aggregated by incubation at 37 ºC for 72 h. Sodium Dodecyl Sulfate (SDS) was also the product of Sigma. Trizol Reagent was purchased from Gibco-BRL (New York, NY, USA). [α-32P]-dATP was from Amersham Pharmacia Biotech (Piscataway, NJ, USA). MMLV reverse transcriptase was from Promega (Madison, WI, USA). Polyclonal integrin αM, c-Jun, and phosphor-c-Jun antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Atlas Mouse 1.2 Expression Arrays and Atlas cDNA Expression Array Kit were provided by Clontech Laboratories. On each array membrane there are 1176 gene cDNA fragments with lengths of 200–600 bp. The name, coordinate, Gene-Bank Number, and other related information for these genes are available on Clontech’s homepage (www.clontech.com).
Animals Male Balb/c mice weighing 22–24 g (Institute of Experimental Animal of Chinese Academy of Medical Sciences, Certificate N
Aggregated Aβ25-35 was icv injected into mice according to the method described by Maurice et al[3]. In brief, a 28-gauge stainless-steel needle was inserted unilaterally 1 mm to the right of the midline point, equidistant from each eye, at an equal distance between the eyes and the ears and perpendicular to the plane of the skull. Aβ25-35 or sterile normal saline at a volume of 5 µL was delivered gradually within 15 s.
Water maze task The Morris water maze test began on d 11 after Aβ-injection as described[7]. The experimental apparatus consisted of a circular water tank (diameter=80 cm) filled to a depth of 15 cm with water maintained at 23±1 ºC. The water was made opaque by adding black ink. A platform (diameter=8 cm) was submerged 0.5 cm below the water surface and placed at the midpoint of one quadrant. Two training trials each day were conducted for 4 consecutive days. On each trial, the mice were placed in the pool at one starting position. They were allowed to swim freely or until they found the platform. The time required to escape onto the hidden platform was recorded. Mice that found the platform were allowed to remain on the platform for 10 s and were then returned to the home cage. If a mouse did not reach the platform within 120 s, it was gently guided to platform by the experimenter, where it remained for 10 s. Statistic comparison of the water maze test between the Aβ-treated and control group was analyzed by two-way analysis of variance (ANOVA).
RNA preparation Mice were killed after the water maze test. Total RNA was extracted from the cerebral cortex with Trizol according to the manufacturer’s instructions. RNA extracts from 10 mice of each group were pooled to provide sufficient material for both cDNA microarray and reverse transcription-PCR analysis. Total RNA was treated with RNase-free DNase at 37 ºC for 20 min to avoid contamination of genomic DNA. The RNA quality and concentration were assessed using agarose gel electrophoresis and spectrophotometric reading.
Probe preparation and hybridization Total RNA (5 µg) was reverse transcribed using reagents provided in the Atlas cDNA Expression Array Kit and radiolabeled with [α-32P]-dATP. The array membranes were prehybridized for 1 h at 68 ºC in ExpressHyb solution containing 100 mg/L of heat-denatured salmon testis DNA. The denatured 32P-labeled cDNA was added to Express Hyb hybridization solution at a final concentration of 4.0 MBq/L and array membranes were hybridized with the labeled cDNA overnight at 68 ºC. The next day, the membranes were washed three times for 30 min with pre-warmed (68 ºC) washing solution 1 [2×SSC (1×SSC is 15 mmol/L sodium chloride, 15 mmol/L sodium citrate), 1% SDS] and once for 30 min with pre-warmed (68 ºC) washing solution 2 (0.1×SSC, 0.5% SDS) with continuous agitation at 68 ºC. After a final wash with 2×SSC at room temperature for 10 min, the membrane was immediately wrapped in plastic film and exposed to X-ray film at -70 ºC.
Detection and analysis Gene expression levels were assessed according to autoradiographic intensity of hybridization signals by software. Only those autoradiograms with similar background and identical signal intensity of housekeeping genes were comparable. Genes that showed an increase or decrease of 2.0-fold or greater were considered as differentially expressed genes. Each group sample was carried out three times, those genes that were all differentially expressed in the three hybridizations were reported as mean±SEM.
Semi-quantitative RT-PCR Expressions of 6 genes were randomly selected to be further confirmed by RT-PCR. Primers were designed using DNA Star software (Perkin Elmer, Norwalk, Conn, USA) and synthesized by Sbs Bio Inc (Beijing, China). Reverse transcription reaction was catalysed by MMLV. PCR was performed for 30 s at 94 ºC, 30 s at 55 ºC, and 1 min at 70 ºC for a cycle, 32 cycles were completed (25 cycles for GAPDH gene), and finally 10 min at 72 ºC. The RT-PCR products were subjected to electrophoresis in 1% agarose gels and quantitative analysis was carried out using the YLN 2000 Gel Analysis System (Yalien, Beijing, China). The gene expression levels were assessed according to the gray ratio value of the target genes and housekeeping gene, GAPDH. The PCR primers used were as follows: GAPDH, forward primer, 5'-CAG CAA CTC CCA CTC TTGCC CCT CCT GTT ATT AT-3'; AP-1, forward primer, 5'-AGGCAGAGA-GGAAGCGCAT-3', reverse primer, 5'-TGGCACCCACTGTTA-ACGTG-3'; NF-κB: forward primer, 5'-GCAGCCTATCACCA-ACTCT-3', reverse primer, 5'-TACTCCTTCTTCTCCACCA-3'; integrin αM, forward primer, 5'-TTAATGACTCTGCGTTT-GCCC-3', reverse primer, 5'- TCATGTCCTTGTACTGCCGCT-3'; ATPase, forward primer, 5'-CCATCTTCTGCACCATC-GTTC-3', reverse primer, 5'- AGACGCCCTGCCTCTTTCAG-3'; nerve growth factor α (NGF α), forward primer, 5'-ACTGG-GTTCTCACAGCTGCC-3', reverse primer, 5'-CGCAGCA-GCATCAGGTCAT-3'; Akt, forward primer, 5'-CCTGTCTCGA-GAGCGTGTGTT-3', reverse primer, 5'-CATAGTGGCACCGT-CCTTGA-3'.
Western blot analysis After decapitation, the cerebral cortex from 5 mice were immediately homogenized in 5 volumes of homogenization buffer [Tris-HCl 50 mmol/L (pH 7.5), NaCl 150 mmol/L, 1% Triton-X 100, 1% sodium deoxycholate, 0.1% SDS]. After sonication and centrifugation at 4 ºC (10 min, 10 000×g), the supernatant was used for determination of protein concentrations, and the equal amounts of total solubilized proteins were eluted by heating with SDSPAGE sample buffer and separated by SDS-polyacrylamide gel electrophoresis. Proteins were then transferred to the nitrocellulose membrane. The membrane was blocked at room temperature with 5% non-fat dry milk in Tris-buffered saline with 0.1% (v/v) Tween-20 (TBST) and incubated with polyclonal integrin αM, c-Jun and phosphor-c-Jun antibody (1:1000) overnight at 4 ºC. After washing, the membrane was incubated with horseradish-labeled secondary antibody for 1 h. After five additional washes in TBST, the membrane was incubated with an enhanced chemiluminescence detecting reagent according to the manufacturer’s protocols and exposed to X-ray film.
Results
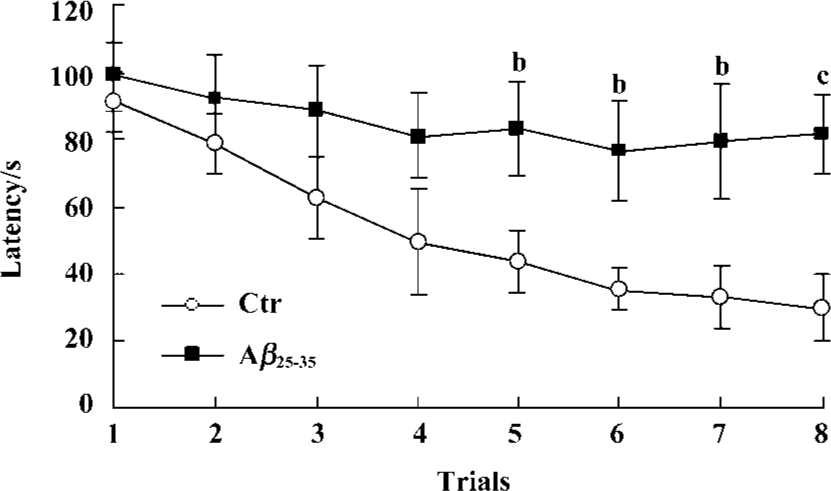
Effects of icv injection of Aβ25-35 on performance of the Morris water maze task Effect of Aβ-injection on learning and memory revealed a significant difference between the two groups. Aβ-injected mice exhibited an obvious learning deficit compared to the saline-injected mice. ANOVA analysis showed a significant decrease in the latency time for the control group during the 8 training trials, but not for the experimental group (Figure 1).
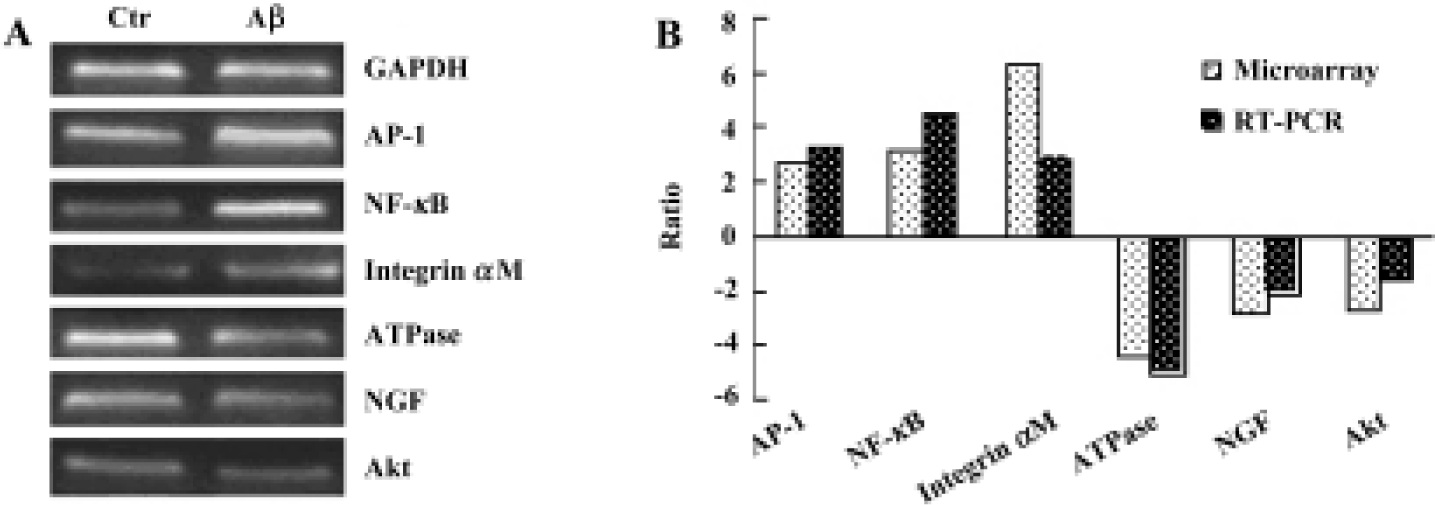
Gene expression analysis of mice cerebral cortex after icv injection of Aβ25-35 The human cerebral cortex serves to control functions such as speech, memory, logical and emotional response, as well as consciousness, interpretation of sensation, coordination of voluntary movement, and these are all greatly involved and damaged in AD. In our study, we used cDNA microarray to investigate the molecular events of neuronal toxicity induced by Aβ25-35 in the cerebral cortex of mice. We compared the gene expression levels in the cerebral cortex of Aβ-injected mice and saline-injected mice. Of 1176 known mouse genes represented on the array, we detected 31 genes differentially expressed in the mice cerebral cortex. Among them 19 genes showed up-regulation in expression after injection of Aβ, including TBX1, NF-κB, AP-1/c-Jun, cadherin, integrin, erb-B2, and FGFR1; while expression of 12 genes were down-regulated, including NGF, glucose phosphate isomerase 1, AT motif binding factor 1, Na+/K+-ATPase, and Akt. All differentially expressed genes were functionally characterized as a diverse spectrum of the biologic process, including transcription factor, cell cycle protein, cytokines, signal transduction, energy metabolism, neurotrophic factor, and so on (Table 1).

Full table
Semi-quantitative RT-PCR To confirm the results of cDNA array, RT-PCR analysis was performed to examine the expression level of 6 genes that were up-regulated or down-regulated in the cerebral cortex of mice. The RT-PCR results indicated that 6 genes showed identical results to that of the microarray (Figure 2).
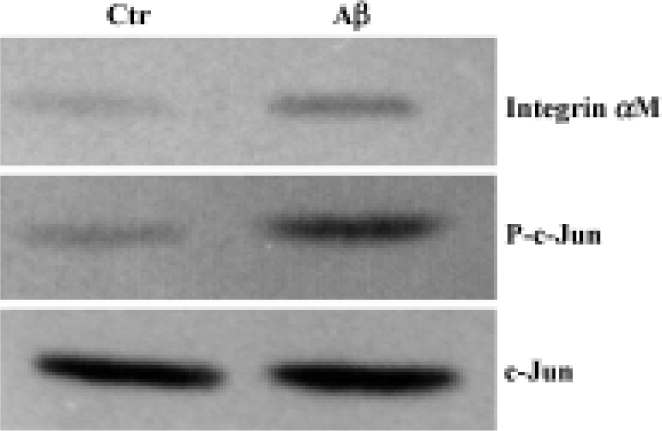
Western blot analysis The expression of integrin αM and activated AP-1/c-Jun of the cerebral cortex were investigated by Western blot analysis. Although total c-Jun was unchanged in intensity between the two groups, the increased expression of integrin αM and phosphorylated c-Jun were found in the cerebral cortex of Aβ-treated mice (Figure 3).
Discussion
Ample experimentation indicates that icv injection of Aβ to rodents can induce learning and memory impairments as well as neurodegeneration in brain areas related to cognitive function[3–5]. This model of Aβ exposure is a useful model in vivo for AD. In the present study, we used the Morris water maze test to detect the effect of aggregated Aβ25-35 injection on the learning and memory of mice. Our results corresponded to previous reports that Aβ25-35 could significantly impair the spacial learning ability and memory of mice.
Microarray provides a promising approach to explore novel molecular and cellular mechanisms that may contribute to some diseases. Gene expression changes in the mice cerebral cortex between icv injection of Aβ25-35 and sterile normal saline were analyzed by using cDNA microarray. In total, 31 genes were differentially expressed, of which 19 genes were increased and 12 were decreased. This implies that only a small fraction of the surveyed genes were affected by Aβ25-35, while most stayed at a stable expression level. The up-regulation or down-regulation of these genes may be responsible for Aβ-induced memory impairment in mice.
In the present study, we found that several genes encoding transcription and chromatin-modifying factors were differentially expressed after icv administration of Aβ25-35. For example, TBX1, transcription factor 21, transcription factor AP-1/c-Jun, and NF-κB showed an increased expression level, while engrailed homeobox protein 1 and Oct-1 were decreased in expression. AP-1 complex is composed of a heterodimer Fos and Jun, the latter is involved in p38/c-Jun N-terminal MAPK pathway (JNK) and considered as a major regulator of both neuronal death and regeneration[8,9]. A number of reports indicate that NF-κB regulates the expression of various genes that play critical roles in apoptosis, viral replication, tumorigenesis, various autoimmune diseases, and inflammation[10,11]. Studies of postmortem brain tissue from patients with AD have revealed increased NF-κB activity in cells involved in the neurodegenerative process. This is consistent with increased p65 immunoreactivity in neurons and astrocytes in the immediate vicinity of amyloid plaques in brain sections from AD patients[12]. It is reported that Aβ can activate NF-κB in the cultured neurons[13], suggesting a molecular mechanism by which Aβ may act during AD pathogenesis. We also noticed that the most dramatic change in gene expression was for the transcript encoding for TBX1, a member of the T-box binding gene family[14]. There are at least 21 members of this gene family. The T-box region, which mediates DNA binding, is highly conserved from Drosophila to humans. Human and mice TBX1 proteins share 98% amino acid identity overall and are identical except for 2 residues within the T-box domain. Expression of human TBX1 in adult and fetal tissues, as determined by Northern blot analysis, were similar to that found in the mice[15]. These transcription factors are involved in the regulation of developmental processes, where they seem to be required for tissue-specific development. In particular, the TBX1 gene is a major genetic determinant in Del22q11.2 syndrome in human beings, which is characterized by a 3-Mb deletion on chromosome 22q11.2, cardiac abnormalities, T-cell deficits, cleft palate facial anomalies, and hypocalcemia[16]. However, there is little known about specific target genes for TBX1 proteins and the relationship between this gene and AD. The function of the most up-regulated gene, TBX1, in the Aβ-injection mice should be further studied.
Integrin αM and integrin αL, two cell adhesion receptors with an Aβ binding site and involved in the uptake of Aβ in neurons and glial cells[17], were observed to be up-regulated in the Aβ-treated mice. Adhesion proteins usually transduce the extracellular signal into cells and change the cell cytoskeleton. It is very possible that Aβ binds to integrin proteins and activates the focal adhesion (FA) proteins, paxillin and focal adhesion kinase (FAK). In this regard, fibrillar Aβ could promote dystrophy by aberrantly activating FA signal transduction cascades that would be involved in Aβ-induced neuronal dystrophy or cell death[18,19]. More-over, the binding of Aβ to integrin proteins would induce sustained activation of MAPK pathways followed by neurite degeneration and hyperphosphorylation of tau proteins[20]. N-cadherin, another cell-adhesion protein, also showed an increase in expression after Aβ-injection. It is a cell surface glycoprotein that mediates calcium-dependent adhesion between neural cells and affects a wide range of cellular processes, including cell adhesion, cell morphogenesis and cell migration.
Two protein tyrosine kinases, erb-B2, and FGF receptor 1, were increased in expression after Aβ-injection. Erb-B2 is the receptor of neuregulin, and overexpression has been demonstrated to activate mitogenic signal transduction and enhance proliferative, prosurvival, and metastatic signals in cell lines. The activity of the MAPK pathway is up-regulated in cells overexpressing erb-B2[21,22]. However, the understanding of the function of erb-B2 in neural cells is very limited. This basic growth factor has pleiotropic effects and plays a regulatory role in angiogenesis, smooth muscle cell growth and wound healing through fibroblast growth factor receptors (FGFR)[23]. FGFR1 activation is followed by intracellular signaling cascades involving Ca2+ channels, phospholipase C, src family kinases, mitogen-activated kinases and the Grb2/Sos signaling complex[24,25]. An increase of FGFR2 in brain neurons and retinal pigment epithelial cells can lead to the activation of L-type channels of neuroendocrine and induce gene expression through the phosphorylation of CREB (cyclic AMP-responsive element binding protein)[26]. Whether the up-regulation of neuroendocrine protein 1 resulted from the increase of FGFR 1 in our study still needs to be studied further.
It is well known that Aβ can impair mitochondria energy metabolism and selectively inhibit Na+/K+-ATPase activity both in vivo and in vitro. From our present results, we found that two Na+/K+-ATPase related genes, AT motif-binding factor 1 (ATBF1), and Na+/K+-ATPase 2β2, were down-regulated. This is consistent with previous studies. ATBF1 is a large transcription factor protein containing four homeo-domains and 23 zinc finger motifs. It also includes several motifs that were found in ATPases[27]. The ATPase activity has been reported to be associated with ATBF1 activity in a unique DNA/RNA-dependent manner that requires both homeodomain and zinc finger motifs[28].
Interleukin 3 receptor has been reported to have a neurotrophic effect on central cholinergic neurons[29]. Down-regulation of this gene may be responsible for cholinergic dysfunction induced by Aβ. Another two growth factors, NGF and glucose phosphate isomerase 1, were also down-regulated. A large number of reports have shown that these neurotrophic factors have beneficial effects on neural cell survival both in vivo and in vitro.
The expression of an apoptosis associated protein, Akt, decreased in the Aβ-injected mice. This protein can protect cells from a variety of death-promoting insults in several different cell types, including Aβ toxicity[30]. Activated Akt would lead to phosphorylation of the proapoptotic protein Bad, stimulate Bcl-2 and Bax expression, decrease the interaction of Bad with Bcl-xL, increase the Bcl-xL/Bad ratio, eventually promoting cell survival[31].
In summary, we observed extensive activation of transcription factors and the signal transduction system, which may result in neuron apoptosis, energy metabolism and calcium ion homeostasis dysfunction, cell adhesion and neuronal dystrophy. A number of the candidate genes we describe have not been linked to AD in previous publications, and define novel injury pathways warranting further studies. The genes found from the cDNA microarray provide some clues for pursuing a more complete understanding of the molecular mechanism of Aβ-induced neural degeneration and memory impairment.
References
- Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 1998;21:479-505.
- Cummings BJ, Pike CJ, Shankle R, Cotman CW. β-Amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging 1996;17:921-33.
- Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered β-amyloid peptides involves cholinergic dysfunction. Brain Res 1996;706:181-93.
- Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, Nabeshima T. Protective effects of idebenone and α-tocopherol on β-amyloid-(1-42)-induced learning and memory deficits in rats: implication of oxidative stress in β-amyloid-induced neurotoxicity in vivo. Eur J Neurosci 1999;11:83-90.
- Flood JF, Morley JE, Roberts E. Amnestic effects in mice of four synthetic peptides homologous to amyloid β protein from patients with Alzheimer disease. Proc Natl Acad Sci USA 1991;88:3363-6.
- Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature 2000;405:827-36.
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 1984;11:47-60.
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 1999;21:326-9.
- Crocker SJ, Lamba WR, Smith PD, Callaghan SM, Slack RS, Anisman H, et al. c-Jun mediates axotomy-induced dopamine neuron death in vivo. Proc Natl Acad Sci USA 2001;98:13385-90.
- Baeuerle PA, Baichwal VR. NF-κB as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv Immunol 1997;65:111-37.
- Baichwal VR, Baeuerle PA. Activate NF-κB or die? Curr Biol 1997;7:R94-6.
- O’Neill LA, Kaltschmidt C. NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 1997;20:252-8.
- Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors α and β protect neurons against amyloid β-peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA 1995;92:9328-32.
- Papaioannou VE, Silver LM. The T-box gene family. Bioessays 1998;20:9-19.
- Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, et al. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics 1997;43:267-77.
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003;362:1366-73.
- Bi X, Gall CM, Zhou J, Lynch G. Uptake and pathogenic effects of amyloid beta peptide 1-42 are enhanced by integrin antagonists and blocked by NMDA receptor antagonists. Neuroscience 2002;112:827-40.
- Berg MM, Krafft GA, Klein WL. Rapid impact of β-amyloid on paxillin in a neural cell line. J Neurosci Res 1997;50:979-89.
- Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: involvement of Src family protein kinases. J Neurosci 2002;22:10-20.
- Anderson KL, Ferreira A. α1 integrin activation: a link between β-amyloid deposition and neuronal death in aging hippocampal neurons. J Neurosci Res 2004;75:688-97.
- Wen D, Peles E, Cupples R, Suggs SV, Bacus SS, Luo Y, et al. Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 1992;69:559-72.
- Tari AM, Lopez-Berestein G. Serum predominantly activates MAPK and Akt kinases in EGFR- and erbB2-over-expressing cells, respectively. Int J Cancer 2000;86:295-7.
- Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev 1997;18:26-45.
- Koike H, Saito H, Matsuki N. Effect of fibroblast growth factors on calcium currents in acutely isolated neuronal cells from rat ventromedial hypothalamus. Neurosci Lett 1993;150:57-60.
- Williams EJ, Furness J, Walsh FS, Doherty P. Characterization of the second messenger pathway underlying neurite outgrowth stimulated by FGF. Development 1994;120:1685-93.
- Rosenthal R, Thieme H, Strauss O. Fibroblast growth factor receptor 2 (FGFR2) in brain neurons and retinal pigment epithelial cells act via stimulation of neuroendocrine L-type channels (Cav1.3). FASEB J 2001;15:970-7.
- Miura Y, Tam T, Ido A, Morinaga T, Miki T, Hashimoto T, et al. Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. J Biol Chem 1995;270:26840-8.
- Kawaguchi M, Miura Y, Ido A, Morinaga T, Sakata N, Oya T, et al. DNA/RNA-dependent ATPase activity is associated with ATBF1, a multiple homeodomain-zinc finger protein. Biochim Biophys Acta 2001;1550:164-74.
- Tabira T, Chui DH, Fan JP, Shirabe T, Konishi Y. Interleukin-3 and interleukin-3 receptors in the brain. Annal NY Acad Sci 1998;840:107-16.
- Martin D, Salinas M, Lopez-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Aβ (25-35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem 2001;78:1000-8.
- Seo JH, Rah JC, Choi SH, Shin JK, Min K, Kim HS, et al. α-Synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J 2002;16:1826-8.