
Small guanine nucleotide-binding protein Rho and myocardial function
Introduction
RhoA, along with RhoB, RhoC, Rac1, Rac2, Cdc42, RhoG, and TC10, belongs to the Ras family of small GTP-binding proteins. RhoA and Rho-kinase (ROCK), a downstream target protein of small GTP-binding protein Rho, are known to regulate a wide variety of cellular processes such as changes in cell morphology, cell motility, focal adhesions, light chain phosphorylation, and cytokinesis[1,2]. Inhibition of the RhoA/ROCK cascade has been demonstrated to elicit beneficial effects on function of both heart tissues and vasculature. RhoA is believed to play a key role in cell growth, myofibrillar assembly, cardiac hypertrophy, hypertension, vascular smooth muscle cell proliferation and migration in response to heterotrimeric G protein receptor stimulation and to mechanical strain or tyrosine kinase growth factors[3–8]. ROCK has been implicated to mediate the angiotensin II (Ang II)-induced hypertrophic responses of vascular smooth muscle cells and hypertensive vascular diseases in hypertension[9,10]. In addition to cardiovascular diseases, RhoA and ROCK also participate in normal physiological processes such as pregnancy and early heart development. The mRNA expression of RhoA and two types of ROCK (α and β) was found to be elevated in the pregnant myometrium[11], which may be responsible for the augmented myometrial contractility during pregnancy. Using the small interfering RNAs (siRNA) technique, it was reported that disruption of RhoA expression resulted in absence of heart tube fusion and abnormal head development in chick embryos, indicating the importance of RhoA for normal embryogenesis and early heart development[12]. Recent studies have indicated that inhibition of the RhoA-ROCK signal pathway may be a potential target for a number of cardiovascular diseases including hypertension, atherosclerosis, diabetes and hypertrophic heart failure[13]. This review will summarize the role of RhoA and ROCK in heart morphology and function, with a special focus on the pathogenesis of heart diseases associated with abnormal RhoA and ROCK signaling. As certain factors such as peripheral vascular resistance may affect cardiac afterload and subsequently cardiac contractile function, the effect of RhoA-ROCK on cardiac morphology and function appears to be more complex than originally thought and requires more intensive research.
Role of RhoA in cardiac hypertrophy Cardiac hypertrophy is a cardiac physiological adaptation in response to pressure or volume overload. However, after a prolonged period of time, this initial adaptive response becomes maladaptive, thus switching the heart from a compensated to a decompensated state and ultimately increased cardiac mortality and morbidity. The Gq-RhoA-ROCK pathway, which may be activated by several neurohormonal factors such as angiotensin II, is believed to function as an important signaling pathway for cardiac hypertrophy or transition from left ventricular hypertrophy to heart failure. Activation of RhoA is essential for sensing externally applied force, subsequently relaying onto the actin cytoskeleton leading to translocation of extracellular signal-regulated kinase (ERK) en route to cardiac hypertrophy[14,15]. It is believed that hypertrophy-related gene expression in response to RhoA activation is mediated through cross-talk with β1 integrin signal pathway via an organized actin cytoskeleton[14,15]. In addition, transcription factor GATA-4, a key regulator of cardiac genes, may also be a nuclear mediator of RhoA participating in sarcomere assembly in cardiomyocytes. Both RhoA and GATA-4 are necessary for sarcomeric reorganization in response to hypertrophic stimuli. It has been demonstrated that over-expression of either protein alone is sufficient to trigger sarcomeric reorganization[16].
In a recent study of heart failure model in Dahl salt-sensitive rats fed an 8% NaCl diet from 8 weeks with or without the ROCK inhibitor Y-27632, Satoh and colleagues reported elevated left ventricular mass, cardiac myocyte cross-sectional area, interstitial fibrosis, and contractile dysfunction shown as reduced left ventricular ejection fraction and fractional shortening, as well as prolongation in contraction/relaxation duration associated with increased protein expression of Galphaq and ROCK in the Dahl salt-sensitive, salt intake Y-27632-untreated group. Interestingly, the degree of myocardial hypertrophy was significantly reduced in conjunction with improved contractile function but no change in interstitial fibrosis following Y-27632 treatment in Dahl salt-sensitive, salt intake rats[17]. These results suggest the possibility that the Gq-ROCK signal pathway plays an important role in the process of hypertension-induced left ventricular hypertrophy leading to contractile dysfunction. The notion of a key role for RhoA-ROCK in cardiac hypertrophy received convincing support from transgenic studies using mice overexpressing RhoA or a constitutively-activated RhoA mutant in atria and ventricles[18]. Heterozygotes displayed high premature mortality, ventricular dilatation (without change in mass) and dysfunction, changes in expression of hypertrophic index gene, increases in atrial mass, marked conduction abnormalities, and other signs of heart failure[18]. Nevertheless, the phenotype of cardiac anomalies in these transgenic mice is different from those commonly seen in compensated hypertrophy, thus precluding any firm conclusion to be drawn regarding the role of RhoA in cardiac hypertrophy. The possible role of RhoA in cardiac hypertrophy also received indirect evidence from the work related to 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, or statins. Statins have been shown to inhibit cardiac hypertrophy by cholesterol-independent mechanisms including inhibition of activation of RhoA and Rac1[19]. Since Rac1 is a crucial component of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is a main source for reactive oxygen species (ROS) in the hearts, the fact that statins inhibit Rac1-mediated oxidative stress may contribute to their inhibitory effects on cardiac hypertrophy[19].
RhoA and cardiac electromechanical function Although there have been some indications that inhibition of RhoA-ROCK may benefit cardiac dysfunction under conditions such as heart failure or ischemia-reperfusion cardiac injury[20], the precise role of RhoA-ROCK on cardiac contractile function remains unclear. Using the Dahl salt-sensitive heart failure model, it was demonstrated that inhibition of ROCK with Y-27632 significantly improved cardiac contractile dysfunction in Dahl salt-sensitive high salt intake heart failure model including reconciling reduced in left ventricular ejection fraction and fractional shortening, prolonged duration of contraction and relaxation, associated with lessened cardiac hypertrophy. These authors also found that the upre-gulated proto-oncogene c-fos gene expression, but not that of ERK and p70S6 kinase phosphorylation, under heart failure rats was decreased by inhibiting ROCK, suggesting a differential activation of the Rho-ROCK and the ERK-p70S6 kinase pathways in the failing hearts of Dahl salt-sensitive hypertensive rats[20]. The observation that ROCK inhibition rescues cardiac contractile defects (reduced contractility and prolonged duration of contraction/relaxation) is supported by cardiac function assessment obtained from the RhoA transgenic mice. Cardiac-specific overexpression of RhoA triggered prolongation of action potential duration and reduction in ventricular contractility[18]. In a separate study, cardiac-specific inhibition of ROCK was achieved by expressing Rho GDIα, an endogenous specific GDP dissociation inhibitor for Rho family proteins, using the α-myosin heavy chain (α-MHC) promoter. Increased expression of Rho GDIα preserved the left ventricular systolic and diastolic function both before and after the development of cardiac hypertrophy, indicating that Rho GTPases may not be required for maintenance of ventricular contractile function under basal physiological conditions[21]. The RhoA-ROCK also participates in the function of cardiac conducting system. Electrocardiography and intracardiac electrophysiological evidence suggest first- and second-degree atrioventricular (AV) block in cardiac-specific Rho GDIα (which inhibits Rho GTPase) transgenic hearts at 1 and 4 weeks of age, respectively, prior to the development of cardiac hypertrophy[21]. These results suggest that the RhoA-ROCK signal cascade is necessary for cardiac electrical conduction under normal physiological conditions. Further examination revealed that expression of connexin 40 was significantly decreased from 1 to 4 weeks of age in the Rho GDIα transgenic heart, which may contribute, at least in part, to the conduction defects in the Rho GDIα transgenic mice[21].
RhoA, hypertension, and cardiac afterload Regulation of vascular smooth muscle cell contractile state is critical for the maintenance of vascular tone. The RhoA-ROCK signal cascade may affect heart function indirectly through regulation of peripheral vascular resistance. The RhoA-ROCK pathway has been demonstrated to play an important role in a wide variety of cell functions in the vasculature including actin cytoskeleton organization and vascular smooth muscle contraction. The RhoA-ROCK pathway is constitutively active in a number of organs including vascular smooth muscle under physiological and pathophysiological conditions[22]. It is believed that upregulated RhoA-ROCK signal cascade promotes cytosolic Ca2+ sensitization and vascular tension in smooth muscles, leading to enhanced peripheral vascular resistance, vascular tone and subsequently hypertension[22,23]. Myosin phosphatase is the key enzyme responsible for myosin light chain (MLC) dephosphorylation that regulates smooth muscle cell contraction. Vasoconstrictors and vasodilators are expected to inhibit or promote the activity of myosin phosphatase, respectively. It was indicated that G-protein-coupled receptor agonists might inhibit the activity of myosin phosphatase leading to vasoconstriction through activation of RhoA-ROCK. However, nitric oxide (NO) may activate myosin phosphatase, leading to vasorelaxation through cGMP-dependent protein kinase. It has been postulated that RhoA regulates vascular smooth muscle contraction through interaction with myosin phosphatase-Rho interacting protein, a likely new member of the myosin phosphatase controlling myosin light chain dephosphorylation[24].
In addition to the peripheral effect, blockade of the RhoA-ROCK signal pathway in nucleus tractus solitarii (NTS) of brain stem decreased blood pressure, heart rate, and renal sympathetic nerve activity in both Wistar-Kyoto (WKY) rats and spontaneously hypertensive rats (SHR), suggesting a role of the RhoA-ROCK signal cascade in central regulation of blood pressure[7]. Interestingly, the magnitude of blood pressure, heart rate, and renal sympathetic nerve activity drop was much greater in SHR than in WKY rats. Furthermore, membrane RhoA expression and ROCK activity in NTS were enhanced in SHR compared with WKY rats, confirming the contribution of the NTS RhoA-ROCK pathway to blood pressure regulation via sympathetic nervous system[7]. Additional work from the same group suggested that activation of the RhoA-ROCK signal pathway might contribute to neurogenic hypertensive mechanisms caused by chronic inhibition of NO synthesis[8]. Chronic inhibition of NO synthesis by the NO synthase (NOS) inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) is known to trigger the onset of hypertension, which was alleviated by the ROCK inhibitor, Y-27632. Expression of membranous RhoA and phosphorylation of the target proteins of ROCK, the ERM (ezrin, radixin, moesin) family members, was significantly greater in the L-NAME-treated group than control group, indicating that activation of the RhoA-ROCK signal pathway contributes to neurogenic hypertension triggered by chronic NOS inhibition[8].
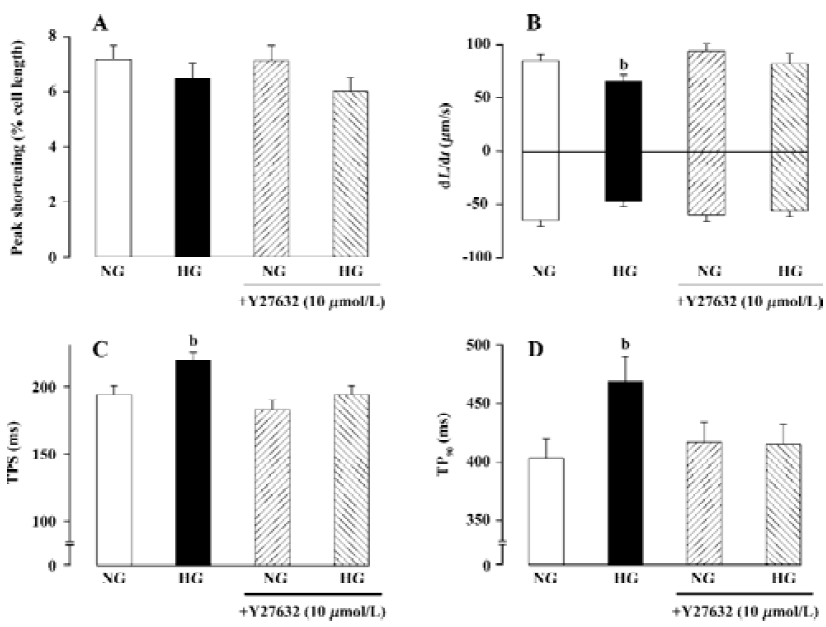
RhoA and diabetic cardiovascular complications Prolonged contraction and relaxation are hallmarks of diabetic cardiomyopathy, one of the most devastating complications in diabetes[25,26]. Studies from our laboratory depicted prolonged duration of contraction and relaxation associated with normal contractility and maximal rate of contraction and relaxation in diabetes[25–27]. Although several mechanisms have been postulated for the diabetes-related mechanical defects such as diabetes-induced myosin isozyme switch (from the fast type V1 to the slow type V3), impaired function of sarco(endo) plasmic reticulum Ca2+-ATPase (SERCA) and Na+/Ca2+ exchanger, as well as reduced myofilament Ca2+ sensitivity [26,28–30], the signaling mechanisms responsible for these cellular alterations remain poorly defined. Recently, we reported up-regulation of cardiac RhoA signaling in diabetic hearts[27]. RhoA expression has been demonstrated to be up-regulated in arteries from aged, hypertensive, or diabetic rats[27,31–33]. The dynamics of RhoA gene expression is sparsely documented although binding of transcription factor cAMP-responsive element binding (CREB) protein/ activating transcription factor-1 (ATF-1) has been shown to increase the RhoA promoter activity[34]. Hyperglycemia and reactive oxygen species, which often accompany hyper-glycemia, have been shown to increase nuclear CREB activity through a p38 MAP kinase-dependent pathway[35,36]. Elevated oxidative stress in diabetic heart and enhanced p38 MAP kinase activation were observed in a preliminary study (Ren and colleagues, unpublished data), which may be related to enhanced expression of RhoA and activity of ROCK. Although there is little evidence depicting a direct regulation of RhoA signaling in cardiac excitation-contraction coupling, activation of ROCK, the Rho-dependent serine-threonine kinase, has been shown to regulate cardiac contractility and gene expression, probably mediated through the MAP kinase super-family[6,18,20]. One of the likely scenarios that RhoA may participate cardiac excitation-contraction coupling is speculated to be mediated through regulation of K+ channel and action potential duration. Cardiac-specific overexpression of RhoA was reported to prolong action potential duration and to diminish ventricular contractility[18]. The RhoA-induced prolongation of action potential may be related to its ability to interrupt certain voltage-dependent K+ channel(s)[37]. These speculations are supported by our recent observation of concurrent up-regulation of RhoA mRNA/protein and diminished Kv1.2 protein expression in diabetic hearts (Ren and colleagues, unpublished data). Depressed expression and function of voltage-dependent K+ channels have also been implicated in prolonged phase 3 of action potential repolarization and thus lengthened relaxation duration in diabetic hearts[38]. The involvement of RhoA signaling in diabetic heart dysfunction was supported by our finding using the ROCK inhibitor Y27632. Incubation of Y-27632 with high extracellular glucose (25.5 mmol/L) for 12 h significantly reduced the compromised cardiac mechanical function elicited by high glucose toxicity (Figure 1), suggesting that RhoA-ROCK activation may be a permissive step in the development of cardiac mechanical defects in response to high extracellular glucose or diabetic environments.
Our recent study revealed that IGF-1 transgene protected diabetes-induced mechanical dysfunctions in cardiac myocytes[39] in parallel to its action on STZ-induced elevation of RhoA mRNA and protein expression, down-regulation of Kv1.2 channels and activation of p38 MAP kinase (Ren and colleagues, unpublished data). Our results did not favor any involvement of another MAP kinase signal molecule ERK1/2 in diabetes-induced cardiomyocyte dysfunction or the cardiac protective effect of IGF-1 (Ren and colleagues, unpublished data). Collectively, these data suggest that up-regulation of the RhoA-ROCK signal cascade may suppress the expression or function of voltage-dependent K+ channel and p38 MAP kinase, an route to mechanical dysfunction in diabetes manifested as prolonged duration of contraction and relaxation, as well as reduced con-tractility. It is possible that the beneficial effects of IGF-1 on cardiac mechanical defects diabetes may be elicited though suppression of activation of the RhoA-ROCK signal cascade[40,41].
Results from our study suggested likely additional signaling mechanisms from RhoA and p38 MAP kinase (Ren and colleagues, unpublished data). Activation of p38 MAP kinase has been speculated to be down-stream of the ROCK signaling pathway[35]. However, current data failed to display that an enhanced ROCK I/II mRNA expression in diabetic groups, indicating that the activity rather than mRNA expression of ROCK I/II may be involved. Nevertheless, IGF-1 transgene reduced ROCK I/II mRNA, consistent with the notion that IGF-1 may inactivate RhoA signal cascade. The lack of effect on ERK1/2 phosphorylation in response to either diabetes or IGF-1 is somewhat consistent with our earlier report using the high glucose culture system[42].
RhoA and other cardiovascular complications Ample of evidence has demonstrated that the RhoA-ROCK mediated signaling pathway plays an important role in other cardiovascular complications such as ischemia/reperfusion heart injury and ventricular remodeling following myocardial infarction. It was reported that fasudil, a potent ROCK inhibitor, prevented occurrence of myocardial ischemia in patients with microvascular angina[43]. It was found that ischemia/reperfusion upregulated RhoA expression in ischemic myocardium and increased ROCK activity. Inhibition of ROCK with selective ROCK inhibitors protected the heart against ischemia injury and enhanced post-ischemia cardiac function. This cardioprotective effect may be attributed to an anti-apoptotic mechanism of ROCK inhibition since ROCK inhibitors may significantly attenuate the downregula-tion of Bcl-2 expression induced by ischemia/reperfusion injury. An alternative explanation for such cardioprotective property of the ROCK inhibitors may be associated with their ability to suppress ischemia/reperfusion-induced elevation in serum levels of proinflammatory cytokines[44]. Further studies have shown that RhoA may be a novel mediator of adenosine-induced cardiac protection against ischemia. The adenosine A3 receptor mediated anti-ischemic function in the heart appeared to signal selectively through RhoA since overexpression the RhoA-noninteracting mutant caused significant reduction of A3 agonist-induced anti-ischemic effect. It was suggested that RhoA was an important cardiopro-tective signaling molecule that interacted directly to phospholipase D (PLD) in cardiac myocytes[45]. Inhibition of ROCK attenuates production of superoxide, reduces generation of monocyte chemoattractant protein-1 or plasminogen activator inhibitor-1 (PAI-1), and inhibits activation of macrophages, neutrophils, and platelets, all of which are considered essential for inhibition of stress-induced regional inflammatory responses and diminished myocardial ischemia-reperfusion injury[46]. Furthermore, RhoA was found to mitigate the reperfusion-induced change in the shape of cardiac capillary endothelial cells in situ and thereby ameliorate the reperfusion injury[47].
Apart from cardiac ischemia/reperfusion injury, RhoA-ROCK has also been demonstrated to be involved in the process of myocardial infarction. It is generally accepted that inhibition of ROCK directly relaxes vascular smooth muscle and therefore increases regional myocardial blood flow at sites of major coronary artery stenosis by dilating abnormally narrowed or occulted artery. However, a recent study revealed that the infarct-limiting mechanism which involved ROCK inhibition could be independent of either a change in systemic hemodynamics or recruitment of collateral blood flow. Transient accumulation of cAMP was demonstrated in myocardium during ischemic preconditioning and may play a role to limit the size of infarction[48]. Other mechanisms such as down-regulating inflammatory responses mediated by cytokines and chemokines may also be involved in limiting the growth of infarct size[49] although further study is warranted to elucidate the therapeutic value of ROCK inhibitors in the management of myocardial infarction.
Summary and conclusion During the past few years, considerable progress has been made toward understanding the signaling pathways and the function of RhoA-ROCK in cardiovascular systems. These studies have convincingly revealed that RhoA-ROCK are versatile signaling molecules regulating diverse cellular functions including cytoskeletal rearrangement[50], Ca2+ sensitization[51], cytokinesis[52] and lineage commitment as well as fate of stem cells[53]. However, much remains to be learned about the detailed regulation mechanisms of RhoA-ROCK as well as its downstream signaling targets. It is imperative that future efforts be directed toward better defining and characterizing the signal pathways regulated by RhoA-ROCK in cardiovascular systems. Such efforts will likely yield new molecular targets for the RhoA-ROCK signal cascade and ultimately more effective therapies for preventing or ameliorating cardiovascular diseases such as cardiac hypertrophy, hypertension, diabetes and atherosclerosis through either pharmacologically or genetic modulation of RhoA-ROCK regulated signaling.
Acknowledgement
The Ren laboratory has been supported by grants from the American Diabetes Association, the American Heart Association, the National Institute of Health, the North Dakota Max Baer Heart Fund, and a University of Wyoming Research Grant.
References
- Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, et al. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science 1997;275:1308-11.
- Yasui Y, Amano M, Nagata K, Inagaki N, Nakamura H, Saya H, et al. Roles of Rho-associated kinase in cytokinesis: mutations in Rho-associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol 1998;143:1249-58.
- Hoshijima M, Sah VP, Wang Y, Chien KR, Brown JH. The low molecular weight GTPase Rho regulates myofibril formation and organization in neonatal rat ventricular myocytes: involvement of Rho kinase. J Biol Chem 1998;273:7725-30.
- Majumdar M, Seasholtz TM, Goldstein D, de Lanerolle P, Brown JH. Requirement for Rho-mediated myosin light chain phosphorylation in thrombin-stimulated cell rounding and its dissociation from mitogenesis. J Biol Chem 1998;273:10099-106.
- Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res 1999;84:1186-93.
- Clerk A, Sugden PH. Small guanine nucleotide-binding proteins and myocardial hypertrophy. Circ Res 2000;86:1019-23.
- Ito K, Hirooka Y, Sakai K, Kishi T, Kaibuchi K, Shimokawa H, et al. Rho/Rho-kinase pathway in brain stem contributes to blood pressure regulation via sympathetic nervous system: possible involvement in neural mechanisms of hypertension. Circ Res 2003;92:1337-43.
- Ito K, Hirooka Y, Kishi T, Kimura Y, Kaibuchi K, Shimokawa H, et al. Rho/Rho-kinase pathway in the brainstem contributes to hypertension caused by chronic nitric oxide synthase inhibition. Hypertension 2004;43:156-62.
- Yamakawa T, Tanaka S, Numaguchi K, Yamakawa Y, Motley ED, Ichihara S, et al. Involvement of Rho-kinase in angiotensin II-induced hypertrophy of rat vascular smooth muscle cells. Hypertension 2000;35:313-8.
- Mukai Y, Shimokawa H, Matoba T, Kandabashi T, Satoh S, Hiroki J, et al. Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. FASEB J 2001;15:1062-4.
- Nirro N, Nishimura J, Sakihara C, Nakano H, Kanaide H. Up-regulation of Rho A and Rho-kinase mRNAs in the rat myometrium during pregnancy. Biochem Biophys Res Commun 1997;230:356-9.
- Kaarbo M, Crane DI, Murrell WG. RhoA is highly up-regulated in the process of early heart development of the chick and important for normal embryogenesis. Dev Dyn 2003;227:35-47.
- Hu E, Lee D. Rho kinase inhibitors as potential therapeutic agents for cardiovascular diseases. Curr Opin Investig Drugs 2003;4:1065-75.
- Wei L, Wang L, Carson JA, Agan JE, Imanaka-Yoshida K, Schwartz RJ. beta1 Integrin and organized actin filaments facilitate cardiomyocyte-specific RhoA-dependent activation of the skeletal alpha-actin promoter. FASEB J 2001;15:785-96.
- Kawamura S, Miyamoto S, Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. J Biol Chem 2003;278:31111-7.
- Charron F, Tsimiklis G, Arcand M, Robitaille L, Liang Q, Molkentin JD, et al. Tissue-specific GATA factors are transcriptional effectors of the small GTPase RhoA. Genes Dev 2001;15:2702-19.
- Satoh S, Ueda Y, Koyanagi M, Kadokami T, Sugano M, Yoshikawa Y, et al. Chronic inhibition of Rho kinase blunts the process of left ventricular hypertrophy leading to cardiac contractile dysfunction in hypertension-induced heart failure. J Mol Cell Cardiol 2003;35:59-70.
- Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, Ross J Jr, et al. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest 1999;103:1627-34.
- Nakagami H, Liao JK. Statins and myocardial hypertrophy. Coron Artery Dis 2004;15:247-50.
- Kobayashi N, Horinaka S, Mita S, Nakano S, Honda T, Yoshida K, et al. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res 2002;55:757-67.
- Wei L, Taffet GE, Khoury DS, Bo J, Li Y, Yatani A, et al. Disruption of Rho signaling results in progressive atrioventricular conduction defects while ventricular function remains preserved. FASEB J 2004;18:857-9.
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 2003;83:1325-58.
- Surks HK, Richards CT, Mendelsohn ME. Myosin phosphatase-Rho interacting protein: a new member of the myosin phosphatase complex that directly binds RhoA. J Biol Chem 2003;278:51484-93.
- Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol Heart Circ Physiol 1997;272:H148-H158.
- Ren J, Bode AM. Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol Heart Circ Physiol 2000;279:H238-44.
- Duan J, Zhang HY, Adkins SD, Ren BH, Norby FL, Zhang X, et al. Impaired cardiac function and IGF-I response in myocytes from calmodulin-diabetic mice: role of Akt and RhoA. Am J Physiol Endocrinol Metab 2003;284:E366-76.
- Dillmann WH. Diabetes and thyroid-hormone-induced changes in cardiac function and their molecular basis. Annu Rev Med 1989;40:373-94.
- Hofmann PA, Menon V, Gannaway KF. Effects of diabetes on isometric tension as a function of [Ca2+] and pH in rat skinned cardiac myocytes. Am J Physiol Heart Circ Physiol 1995;269:H1656-63.
- Chattou S, Diacono J, Feuvray D. Decrease in sodium-calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol Scand 1999;166:137-44.
- Miao L, Calvert JW, Tang J, Parent AD, Zhang JH. Age-related RhoA expression in blood vessels of rats. Mech Ageing Dev 2001;122:1757-70.
- Seasholtz TM, Zhang T, Morissette MR, Howes AL, Yang AH, Brown JH. Increased expression and activity of RhoA are associated with increased DNA synthesis and reduced p27(Kip1) expression in the vasculature of hypertensive rats. Circ Res 2001;89:488-95.
- Massey AR, Miao L, Smith BN, Liu J, Kusaka I, Zhang JH, et al. Increased RhoA translocation in renal cortex of diabetic rats. Life Sci 2003;72:2943-52.
- Sauzeau V, Rolli-Derkinderen M, Marionneau C, Loirand G, Pacaud P. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem 2003;278:9472-80.
- Singh LP, Andy J, Anyamale V, Greene K, Alexander M, Crook ED. Hexosamine-induced fibronectin protein synthesis in mesangial cells is associated with increases in cAMP responsive element binding (CREB) phosphorylation and nuclear CREB: the involvement of protein kinases A and C. Diabetes 2001;50:2355-62.
- Ichiki T, Tokunou T, Fukuyama K, Iino N, Masuda S, Takeshita A. Cyclic AMP response element-binding protein mediates reactive oxygen species-induced c-fos expression. Hypertension 2003;42:177-83.
- Cachero TG, Morielli AD, Peralta EG. The small GTP-binding protein RhoA regulates a delayed rectifier potassium channel. Cell 1998;93:1077-85.
- Shimoni Y, Firek L, Severson D, Giles W. Short-term diabetes alters K+ currents in rat ventricular myocytes. Circ Res 1994;74:620-8.
- Norby FL, Aberle NS 2nd, Kajstura J, Anversa P, Ren J. Transgenic overexpression of insulin-like growth factor I prevents streptozotocin-induced cardiac contractile dysfunction and beta-adrenergic response in ventricular myocytes. J Endocrinol 2004;180:175-82.
- Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 2001;50:1414-24.
- Norby FL, Wold LE, Duan J, Hintz KK, Ren J. IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol Endocrinol Metab 2002;283:E658-66.
- Ren J, Duan J, Hintz KK, Ren BH. High glucose induces cardiac insulin-like growth factor I resistance in ventricular myocytes: role of Akt and ERK activation. Cardiovasc Res 2003;57:738-48.
- Mohri M, Shimokawa H, Hirakawa Y, Masumoto A, Takeshita A. Rho-kinase inhibition with intracoronary fasudil prevents myocardial ischemia in patients with coronary microvascular spasm. J Am Coll Cardiol 2003;41:15-9.
- Bao W, Hu E, Tao L, Boyce R, Mirabile R, Thudium DT, et al. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res 2004;61:548-58.
- Mozzicato S, Joshi BV, Jacobson KA, Liang BT. Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J 2004;18:406-8.
- Shimokawa H. Rho-kinase as a novel therapeutic target in treatment of cardiovascular diseases. J Cardiovasc Pharmacol 2002;39:319-27.
- Glyn MC, Lawrenson JG, Ward BJ. A Rho-associated kinase mitigates reperfusion-induced change in the shape of cardiac capillary endothelial cells in situ. Cardiovasc Res 2003;57:195-206.
- Sanada S, Asanuma H, Tsukamoto O, Minamino T, Node K, Takashima S, et al. Protein kinase A as another mediator of ischemic preconditioning independent of protein kinase C. Circulation 2004;110:51-7.
- Hattori T, Shimokawa H, Higashi M, Hiroki J, Mukai Y, Tsutsui H, et al. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation 2004;109:2234-9.
- Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S. Coordination of microtubules and actin cytoskeleton by the Rho effector mDial. Nature Cell Biol 2001;3:8-14.
- Somlyo AV. New roads leading to Ca2+ sensitization. Circ Res 2002;91:83-4.
- Glotzer M. Cytokinesis: progress on all fronts. Curr Opin Cell Biol 2003;15:684-90.
- McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 2004;6:483-95.